S. J. BELL & D. R. FORSDYKE
Journal of Theoretical Biology (1999) 197, 51-61.
[With the permission of the copyright holder, Academic Press]
3. Accounting in Herpes simplex and Vaccinia Viruses
|
Summary. Chargaff's first parity rule (%A=%T and %G=%C) is explained by the Watson-Crick model for duplex DNA in which complementary base pairs form individual accounting units. Chargaff's second parity rule is that the first rule also applies to single strands of DNA. The limits of accounting units in single strands were examined by moving windows of various sizes along sequences and counting the relative proportions of A and T (the W bases), and of C and G (the S bases). Shuffled sequences account, on average, over shorter regions than the corresponding natural sequence. For an E. coli segment, S base accounting is, on average, contained within a region of 10 kb, whereas W base accounting requires regions in excess of 100 kb. Accounting requires the entire genome (190 kb) in the case of vaccinia virus, which has an overall "Chargaff difference" of only 0.086% (i.e. only one in 1162 bases does not have a potential pairing partner in the same strand). Among the chromosomes of Saccharomyces cerevisiae, the total Chargaff differences for the W bases and for the S bases are usually correlated. In general, Chargaff differences for a natural sequence and its shuffled counterpart diverge maximally when 1 kb sequence windows are employed. This should be the optimum window size for examining correlations between Chargaff differences and sequence features which have arisen through natural selection. We propose that Chargaff's second parity rule reflects the evolution of genome-wide stem-loop potential as part of short and long range accounting processes which work together to sustain the integrity of various levels of information in DNA. |
|
When the base composition of natural duplex DNA is determined it is found that the quantities of A and T are equal and the quantities of C and G are equal. This is Chargaff's famous first parity rule (Chargaff, 1951). If a long DNA duplex is cut into two and the base composition of each part determined, the rule is found to hold precisely for the two parts, as for the duplex of origin. This division of the duplex can be continued down to individual bases (pairing with their complementary bases on the opposite strand of the duplex). Again Chargaff's parity rule is obeyed precisely (Watson & Crick, 1953). Disregarding nearest- neighbour influences (Turner, 1996), single base pairs can be regarded as fundamental "accounting units". The summation of these individual accounting units results in the precise A=T and C=G equivalences of duplex DNA sequences. That the equivalences have arisen, and are maintained, because they are of adaptive value to an organism, is not in doubt (Bernstein & Bernstein, 1991). Chargaff's second parity rule is that, to a close approximation, the first rule equivalences also apply to individual single-strands taken from natural duplex DNA molecules. The possible existence of a second rule became evident in the 1960s (Karkas et al., 1968; Chargaff, 1979). Three decades later recognition is increasing (Prabhu, 1993; Forsdyke, 1995c), but stochastic, rather than adaptive, explanations are emphasized (Lobry, 1995; Sueoka, 1995). This may be mistaken. The equal proportions of males and females in most large populations may appear as merely the result of the chance flipping of the sexual coin. However, the ratio is fixed by powerful selective forces which militate against disparities (Darwin, 1871; Fisher, 1958). Equal proportions can be an evolutionarily stable strategy (Smith, 1989). The second rule is particularly apparent when long sequences are examined. For example, the base composition of the "top" strand of chromosome III of Saccharomyces cerevisiae (Oliver et al., 1992), is 98212 (A), 95572 (T), 62125 (C), and 59432 (G). A and T differ by only 2640 bases, and C and G differ by only 2693 bases. Only 1.4% of the W bases (A and T, which pair weakly) are not accounted for by a potential pairing partner. Only 2.2% of the S bases (C and G, which pair strongly) are not accounted for by a potential pairing partner. It appears that there has been some sort of accounting so that the overall "Chargaff difference" for the chromosome is only 1.7%. Is this a function of the whole chromosome (i.e., is the whole chromosome one single accounting unit), or are there smaller accounting units which, when summed, generate this value? The accounting is between A and T, and between C and G, not between A and C (the M [amino] bases), or between T and G (the K [keto] bases). Thus, an accounting process by which Chargaff differences in single strands of DNA are kept small might involve Watson-Crick base-pairing as in the case of duplex DNA. It is known that supercoiled duplex DNA can extrude stem-loops (Murchie et al., 1992), and that there has been an evolutionary pressure on base order favouring the development of extensive stem-loop potential in genomes (Forsdyke, 1995a-d; 1996a,b; 1998). This may derive from the role of "kissing" interactions between complementary loops in the homology search preceding meiotic recombination (Crick, 1971; Kleckner & Weiner 1993; Rocco & Nicolas, 1996). Since efficient recombination would be evolutionarily advantageous (Bernstein & Bernstein, 1991), mutations which improve the ability of DNA to act as a recombination substrate (i.e., mutations favouring the evolution of genome-wide stem-loop potential), would have been accepted. By virtue of the stems in stem-loop structures, there would then be a tendency for there to be equal proportions of A and T, and of C and G, in single strands of DNA. Thus, base pairing in stems provides one possible level of accounting, which would be localized to the region of stem-loop extrusion. It seems unlikely that this relatively short range process could alone explain the precision of single-strand accounting. Base pairing between complementary loops (Tomizawa, 1984; Eguchi et al., 1991), which might occur very efficiently between cis-oriented sequences within one chromosome (Jinks-Robertson et al., 1993), and might operate over long genomic distances (Engels et al., 1994; Henikoff, 1997), might provide another level of accounting. Chargaff's second rule might apply to long genomic segments because of the summation of underlying primary accounting processes involving both stems (short-range accounting) and loops (long-range accounting). These processes might operate over distinct domains ("accounting subdomains") of the segments. If one counted bases in a sequence window which happened to correspond to a subdomain, then Chargaff differences should approach a minimum. If one then moved the window so that it was centered at the intersection of two subdomains, the Chargaff differences should approach a maximum. Thus, one should be able to determine the limits of accounting subdomains by moving a window along sequences and counting the bases in each window. Smithies et al., (1981) have provided evidence for accounting domains as so defined. These studies were recently extended by Lobry (1995; 1996a, b). However, the choice of window size was arbitrary. We here present studies in which window sizes have been varied. We are concerned with the precision of Chargaff's second rule, contributed to both by adaptive and by stochastic factors, and the length of DNA needed to achieve that precision.
In the following paper we report that the determined optimum window size actually is optimum for demonstrating such correlations; indeed, deviations from Chargaff's second rule correlate with transcription direction (Bell & Forsdyke, 1998).
|
|
Among the first long sequences obtained as part of the E. coli genome project were two contiguous sequences spanning the 0-4.1 min region of the single E. coli chromosome. These were GenBank sequences ECO110K (0-2.4 min;Yura et al., 1992), and ECO82K (2.4-4.1 min; Fujita et al., 1994), which were combined to generate a segment which we refer to as ECO193K. Chargaff differences, calculated as described previously (Forsdyke, 1998), were determined for windows both in the natural sequence, and in a reference sequence with the same base composition generated by randomizing base order in the natural sequence using the GCG program SHUFFLE (Gribskov & Devereux, 1991). In shuffled sequences the balance between the quantities of two pairing bases would be expected to resemble that resulting from the tossing of a biased coin for which heads (A or C) would be slightly favoured/disfavoured over tails (T or G), respectively, depending on their relative proportions in the total segment. The base composition of the arbitrarily designated "top" strand of ECO193K is 45,886 (A), 46,938 (T), 48,343 (C), and 52,476 (G). A is slightly disfavoured over T (by 1052 bases), and C is disfavoured over G (by 4133 bases). Only 1.13 % of the W bases, and 4.10 % of the S bases, are not accounted for by a potential pairing partner. Differences should approach these limiting values after many tosses. This is shown in Fig. 1 where average absolute Chargaff differences are plotted against the size of sequence windows. With windows of only 200 nt, high differences would be expected since there would be great statistical fluctuations when base "coins" are "tossed" no more than 200 times. Average absolute differences for both the W bases and the S bases are high when windows are 200 nt. Values for the natural sequence exceed those of the shuffled natural sequence, implying evolutionary pressures on base order favouring the generation and maintenance of Chargaff differences. |
|
|
FIG. 1. Variation of average Chargaff difference values with size of windows in sequence ECO193K (193,643 nt), which was assembled by uniting GenBank segment ECO110K (111,402 nt) with overlapping downstream GenBank segment ECO82K (82,727 nt). Windows of varying size were moved along the sequence in steps of 100 nucleotides, and base compositions were determined in each window. Absolute Chargaff differences were calculated for Fig. 1a as deltaW/W and expressed as a percentage. deltaW is the absolute value of the difference between the number of W bases (deltaW=|A-T|), and W is the sum of the W bases (W=A+T). Absolute Chargaff differences were calculated for Fig. 1b as deltaS/S and expressed as a percentage. deltaS is the absolute value of the difference between the number of S bases (deltaS=|C-G|), and S is the sum of the S bases (S=C+G). Average Chargaff differences for each window size are plotted either as large grey symbols (natural sequence), or as large black symbols (shuffled sequence). Small yellow diamonds refer to the ratio of these values (the average Chargaff difference for the natural sequence divided by the average Chargaff difference for the shuffled sequence). Small red diamonds refer to the difference between these values determined by subtraction. The horizontal dotted lines indicate Chargaff differences for the entire sequence (i.e. the largest possible window, of which there is only one copy). Thus, the total number of windows of a given size varies with sequence length. In a 100 kb sequence there will be 999 windows of 0.2 kb, and one window of 100 kb. |
|
With increasing window size average Chargaff differences for both natural and shuffled sequences decrease in an exponential fashion to approach the value for the entire segment (horizontal dotted lines). Much of the decline in Chargaff difference values is achieved with windows in the 1 to 2 kb range, implying effective local accounting, largely due to statistical factors. Windows of about 3 kb are required for average S base Chargaff differences for the shuffled sequence to approach the theoretical limit. However, windows of about 20 kb are required for average S base Chargaff differences for the natural sequence to approach the limit (Fig. 1b). For the W bases, the natural sequence does not reach the limit even with windows extending to 100 kb. The corresponding shuffled sequence reaches the limit with average windows of about 10 kb (Fig. 1a). These results imply that S and W bases are, to some extent, accounted separately, and that while S base accounting is, on average, contained within a region of 10 kb, W base accounting, on average, requires regions in excess of 100 kb. Values for the natural and shuffled sequences were compared either as a ratio (open yellow diamonds), or by subtraction (filled red diamonds). The size of the window at which Chargaff differences for natural and shuffled sequences diverge maximally depends on the method used. In the case of the W bases the maximum divergence by ratio occurs with 4 kb windows, but the maximum divergence by subtraction is with 1.1 kb windows. In the case of the S bases the divergence by the ratio method is high with 1 kb windows, but reaches a maximum with 1.5 kb windows. By the difference method, the divergence reaches a maximum level at 0.6 kb which is sustained to 1.2 kb.
|
3. Accounting in Herpes simplex and Vaccinia Viruses
| Having examined 193 kilobases, a mere 5% of the 4.2 megabase circular chromosome constituting the entire E. coli genome (Fig. 1), we next looked at two linear viral genomes where the size of the maximum possible accounting unit would be presumed to be no greater than the size of the entire genome. The 152 kb Herpes simplex genome (C+G=68.3% of total bases) has overall Chargaff difference values of 1.01% for the S bases, and 0.39% for the W bases (McGeoch et al., 1988). The 192 kb vaccinia virus genome (C+G=33.4%) has overall values of 0.03% for the S bases and 0.11% for the W bases (Goebel et al., 1990). For this virus only one in 3202 of the S bases does not have a potential pairing partner in the same strand. |
|
|
| FIG. 2. Variation of average Chargaff difference with size of windows in Herpes simplex virus (152,260 nt; GenBank locus HE1CG). Details are as in Fig. 1. |
| Fig. 2 shows the effect of varying window sizes on Chargaff differences for the Herpes simplex virus genome. Accounting for the S bases extends to average windows of around 100 kb where values for the natural and for the shuffled sequence converge. Chargaff differences for the W bases in the natural and shuffled sequences coincide when average windows are 30 kb, even though values for the shuffled sequence do not approach the value for the whole genome until average window sizes are around 70 kb. Thus, in the S base-rich Herpes simplex genome, on average, accounting appears to be complete within a distance less than that of the entire genome. Furthermore, S bases "require" more accounting "room" than the W bases. Using the ratio method, average Chargaff difference values for the natural and shuffled sequences diverge maximally with 1 kb windows (W bases; Fig. 2a), and 2 kb windows (S bases; Fig. 2b), with some subsequent peaks at higher window sizes. Using the subtraction method, the divergence is maximum at the smallest window used (0.1 kb), decreasing progressively thereafter, with some suggestion of a shoulder at 1 kb in the case of the S bases. |
|
|
| FIG. 3. Variation of average Chargaff difference with size of windows in vaccinia virus (191,737 nt; GenBank locus VACCG). Details are as in Fig. 1. |
| The entire W base-rich vaccinia virus genome appears to be one large accounting unit (Fig. 3). Average Chargaff differences for both the S and the W bases do not attain the values of the entire chromosome until the ultimate window (the size of the whole chromosome) is reached. W and S bases have equal "requirements" for accounting "room". In both cases, divergences between Chargaff difference values for natural and shuffled sequences by the subtraction method reach a maximum with windows of about 1 kb. This maximum divergence is sustained to 10 kb windows and then progressively declines at higher windows sizes. By the ratio method, divergences increase progressively, and maxima are attained only at high window sizes. |
4. Accounting in Saccharomyces cerevisiae
|
|
| FIG. 4. Variation of average Chargaff difference (ordinate) with size of windows (abscissa) in chromosome III of Saccharomyces cerevisiae (315,341 nt). Details are as in Fig. 1. |
| We next examined a linear genome segment with a defined natural limit, chromosome III of Saccharomyces cerevisiae (315 kb), which is enriched for the W bases (C+G=38.6%; Oliver et al., 1992). Accounting by the S bases extends to average windows of around 80 kb (Fig. 4b), whereas the W bases have not reached the accounting limit with average window sizes of 100 kb (Fig. 4a). Thus, in this W base-rich genome, the W bases "require" more accounting "room" than the S bases. Divergences between the natural and shuffled sequences reach maxima with 2 kilobase windows (ratio method), but the values for 1 kb windows are quite close to the maximum values. Divergences by the subtraction method are maximal at 0.3 kb (W bases), and 0.4 kb (S bases). |
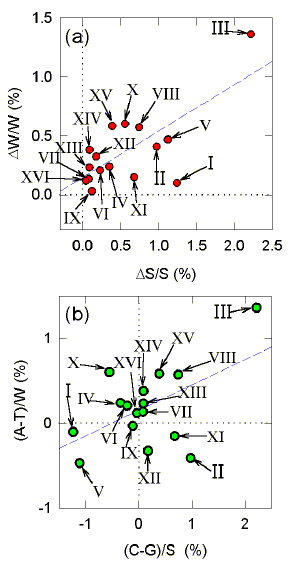
| Chromosome # | A | C | G | T | (C-G)/S (%) | (A-T)/W (%) |
| 1 | 69832 | 44642 | 45762 | 69973 | -1.239 | -0.101 |
| 2 | 249646 | 157412 | 154380 | 251699 | 0.972 | -0.409 |
| 3 | 98212 | 62125 | 59432 | 95572 | 2.215 | 1.362 |
| 4 | 476768 | 289351 | 291363 | 474492 | -0.346 | 0.239 |
| 5 | 176532 | 109828 | 112314 | 178197 | -1.119 | -0.469 |
| 6 | 82928 | 52201 | 52435 | 82584 | -0.224 | 0.208 |
| 7 | 338319 | 207764 | 207449 | 337403 | 0.076 | 0.136 |
| 8 | 174022 | 109094 | 107486 | 172036 | 0.742 | 0.574 |
| 9 | 34340 | 85461 | 85661 | 134423 | -0.117 | -0.031 |
| 10 | 231099 | 142213 | 143801 | 228330 | -0.555 | 0.603 |
| 11 | 206057 | 127713 | 126003 | 206675 | 0.674 | -0.150 |
| 12 | 330586 | 207777 | 207064 | 332744 | 0.172 | -0.325 |
| 13 | 286296 | 176735 | 176433 | 284966 | 0.086 | 0.233 |
| 14 | 241562 | 151651 | 151388 | 239729 | 0.087 | 0.381 |
| 15 | 339395 | 209021 | 207417 | 335449 | 0.385 | 0.585 |
| 16 | 293947 | 180364 | 180507 | 293243 | -0.040 | 0.120 |
| Table 1 shows the base composition of the entire 16-chromosome set of Saccharomyces cerevisiae, together with Chargaff differences. It is noted that chromosome III (the third-smallest chromosome, containing mating type loci), is exceptional in the relatively large size of its Chargaff difference values. The other chromosomes have much lower values, with chromosome XVI (the fifth largest) having the lowest value for the S bases (0.040%); only 1 in every 2500 of the S bases does not have a potential pairing partner. The same chromosome has the third lowest Chargaff difference for the W bases (0.120%), implying highly effective intra-chromosomal accounting for both the S and W bases. Examination of the Chargaff differences of other chromosomes indicates no simple relationship to chromosome length. |
|
|
|
FIG. 5. [Above] Relationship between Chargaff differences for the W bases, and for the S bases, for the 16 individual chromosomes of Saccharomyces cerevisiae. (a) Absolute Chargaff differences. The least-squares regression line (dashed) has a correlation coefficient (r) of 0.72, and a slope of 0.39, which is significantly different from zero (P=0.002). (b) Signed Chargaff differences (using an alphabetical order convention; A-T, C-G). The least-squares regression line has a correlation coefficient (r) of 0.53, and a slope of 0.30, which is significantly different from zero (P=0.034). |
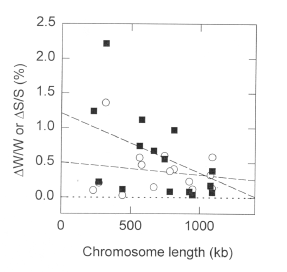
FIG. 6. [Above] Relationship between absolute Chargaff differences and chromosome length for the 16 chromosomes of Saccharomyces cerevisiae. Open circles refer to the W bases, and filled squares refer to the S bases. Dashed lines are the least squares regressions. Only the slope for the absolute Chargaff differences for the S bases is significantly different from zero (r=0.20; P=0.041). |
| Among the chromosomes, absolute Chargaff differences for the W and S bases are usually positively correlated (P = 0.002; Fig. 5a), as are the signed Chargaff differences (P = 0.034 ; Fig. 5b). Since Chargaff differences are expressed as percentages, they should be independent of chromosome size. However, absolute Chargaff differences for the S bases decrease with increasing chromosome size (Fig. 6; P = 0.041). If low Chargaff differences are regarded as the accounting goal, then the small chromosomes would appear to be deviant. |
|
Chargaff's first parity rule for duplex DNA remains valid because evolutionary forces so dictate. Should the base T in the complementary strand opposite an A residue mutate to a C, then the rule is sustained either because the mispairing is corrected before the duplex can divide, or because the C pairs with a G after the division. For a short period of time the rule is violated, but correction is rapid. The pressures for the evolution of this highly efficient "accounting" (error-correcting) process are well understood in terms of DNA structure and function (Watson & Crick, 1953). Organisms which "forget" Chargaff's first rule, are heavily penalized in the course of evolution (Bernstein & Bernstein, 1991). Much less well understood are the evolutionary pressures on organisms not to "forget" Chargaff's second parity rule. Since the base symmetries are the same as in the first rule, it is appropriate to consider the second rule in similar terms as a possible manifestation of processes which have evolved to sustain the integrity of various levels of information in DNA (Forsdyke 1981; 1996b). Application of classical information theory to DNA sequences has indicated the major roles of base composition-dependent and base order-dependent information components, the latter operating primarily at the dinucleotide level (Gatlin, 1972; Sibbald et al., 1989). Most information-theoretic approaches treat DNA sequences as linear strings, without considering the possible information component arising from long range interactions (Sibbald et al., 1989). A need to consider such interactions is apparent in models postulating error-correcting information in DNA (Forsdyke, 1981; Liebovitch et al., 1996) As set out in the Introduction, we do not dismiss single-strand accounting as merely a stochastic phenomenon, but seek an explanation in terms of recent advances in our understanding of the chemistry and biology of stem-loop potential in DNA (Murchie et al., 1992; Forsdyke 1995a-d; 1996 a, b; 1998). The accounting is apparent, not only at the level of single bases as considered here, but also at the levels of the 16 dinucleotides, and of the 64 trinucleotides, and even at higher oligonucleotide levels (Prabhu, 1993). Dinucleotide frequencies appear more fundamental than frequencies of higher oligonucleotides (Nussinov, 1981), consistent with dinucleotide nearest-neighbour stacking interactions being of critical importance for secondary structure (Turner, 1996). In all species examined the frequencies of particular dinucleotides (e.g., AC) closely approximate those of their complements (e.g., GT). This applies both to a natural sequence, and its shuffled counterpart, implying a minimal role of base order (Forsdyke, 1995c). Shuffled natural sequences retain equal proportions of complementary oligonucleotides, but an artificial sequence might be constructed with a low T content, so that AC>GT. Thus, evolutionary forces have acted on base composition to sustain Chargaff's second rule. Absolute Chargaff difference values decrease with increasing window size, especially in the case of shuffled versions (Figs. 1-4). The corresponding natural sequences sustain Chargaff differences at high window sizes, indicating selective evolutionary pressure on base order in this respect. Nevertheless, small Chargaff difference values are achieved at high window sizes. This may not just be a relaxation of selective pressure to allow the operation of stochastic factors. Long range accounting may itself be the result of selective pressures. In the following paper we suggest that stems alone are not sufficient to explain the observed accounting (Bell and Forsdyke, 1999). There should be accounting not only between bases in stems, but also between bases in complementary loops. The latter might be widely separated. Thus, we determined here the range over which single-strand accounting might operate, long range interactions being presumed to depend largely on loop-loop interactions. With minimal assumptions about underlying mechanism, we have shown that the accounting range may extend to many kilobases, and may vary depending on the particular Watson-Crick base-pair studied, and on the organism (Figs. 1-4). At the "macroscopic" scale of the present work, some correlation between W base and S base accounting is evident (Fig. 5). A prima facie case for the existence of long range intra- and inter-chromosomal sensing (and, where necessary, correction), of sequence information is made here in numerical terms. The case can also be made from numerous reports of long range homology recognition phenomena whose adaptive value appears related to error-correction at the level of genes or gene products (Engels et al., 1994; McKee, 1996; Henikoff, 1997). These include enumeration of repeats with subsequent inactivation by mutation (Singer & Selker, 1995), or by methylation (Shemer et al., 1996;), and interphase, mitotic and meiotic recombination (Bernstein & Bernstein, 1991; Jinks-Robertson et al., 1993). The following paper explores the relationship between short and long-range accounting processes at the level of individual genes (Bell & Forsdyke, 1999). |
We thank P. Sibbald for review of the manuscript, J. Gerlach for assistance with computer configuration, L. Russell for technical help, R. Gough, J. Mau and G. Wood for facilitating use of Genetics Computer Group software on the Silicon Graphics computer at the National Research Council, Ottawa, and T. Smith for statistical advice. The work was supported by the Medical Research Council of Canada and Queen's University. [The Oberlin College Computer Science Server (Click Here) was the source of images relating to the Rosetta Stone, which links three ancient information systems.]
BELL, S. J., CHOW, Y. C., HO, J. Y. K. & FORSDYKE, D. R. (1998). Correlation of CHI orientation with transcription indicates a fundamental relationship between recombination and transcription. Gene 216, 285-292.
BELL, S. J. & FORSDYKE, D. R. (1999). Deviations from Chargaff's second parity rule correlate with direction of transcription. J. theor. Biol. 197, 63-76.
BERNSTEIN, C. & BERNSTEIN, H. (1991). Aging, Sex and DNA Repair. San Diego, CA: Academic Press.
CHARGAFF, E. (1951). Structure and function of nucleic acids as cell constituents. Fed. Proc. 10, 654-659.
CHARGAFF, E. (1979). How genetics got a chemical education. Ann. N. Y. Acad. Sci. 325, 345-360.
CRICK, F. (1971). General model for chromosomes of higher organisms. Nature 234, 25-27.
DARWIN, C. (1871). The Descent of Man and Selection in Relation to Sex. pp. 316, London: John Murray.
EGUCHI, Y., ITOH, T. & TOMIZAWA, J. (1991). Antisense RNA. Annu. Rev. Biochem. 60, 631-652.
ENGELS, W. R., PRESTON, C. R. & JOHNSON-SCHLITZ, D. M. (1994). Long range cis preference in DNA homology search over the length of a Drosophila chromosome. Science 263, 1623-1625.
FISHER, R. A. (1958). The Genetical Theory of Natural Selection. pp. 158-160, New York: Dover Publications.
FORSDYKE, D. R. (1981). Are introns in-series error-detecting codes? J. theor. Biol. 93, 861-866.
FORSDYKE, D. R. (1995a). A stem-loop "kissing" model for the initiation of recombination and the origin of introns. Mol. Biol. Evol. 12, 949-958.
FORSDYKE, D. R. (1995b). Conservation of stem-loop potential in introns of snake venom phospholipase A2 genes. An application of FORS-D analysis. Mol. Biol. Evol. 12, 1157-1165.
FORSDYKE, D. R. (1995c). Relative roles of primary sequence and (G+C)% in determining the hierarchy of frequencies of complementary trinucleotide pairs in DNAs of different species. J. Mol. Evol. 41, 573-581.
FORSDYKE, D. R. (1995d). Reciprocal relationship between stem-loop potential and substitution density in retroviral quasispecies under positive Darwinian selection. J. Mol. Evol. 41, 1022-1037.
FORSDYKE, D. R. (1996a). Stem-loop potential in MHC genes: a new way of evaluating positive Darwinian selection. Immunogenetics 43, 182-189.
FORSDYKE, D. R. (1996b). Different biological species "broadcast" their DNAs at different (C+G)% "wavelengths". J. theor. Biol. 178, 405-417.
FORSDYKE, D. R. (1998). An alternative way of thinking about stem-loops in DNA. A case study of the G0S2 gene. J. theor. Biol. 192, 489-504.
FUJITA, N., MORI, H., YURA, T. & ISHIHAMA, A. (1994). Systematic sequencing of the Escherichia coli genome: analysis of the 2.4-4.2 min (110,917-193,643 bp) region. Nucleic Acids Res. 22, 1637-1639.
GATLIN, L. L. (1972). Information Theory and the Living System. New York: Columbia University Press.
GOEBEL, S. J., JOHNSON, G. P., PERKUS, M. E., DAVIS, S. W., WINSLOW, J. P. & PAOLETTI, J. (1990). The complete DNA sequence of vaccinia virus. Virology 179, 247-266.
GRIBSKOV, M. & DEVEREUX, J. (1991). Sequence Analysis Primer. New York, NY: Stockton Press.
HENIKOFF, S. (1997). Nuclear organization and gene expression: homologous pairing and long-range interactions. Curr. Opin. Cell Biol. 9, 388-395.
JINKS-ROBERTSON, S., MICHELITCH, M. & RAMCHARAN, S. (1993). Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 13, 3937-3950.
KARKAS, J. D., RUDNER, R. & CHARGAFF, E. (1968). Separation of B. subtilis DNA into complementary strands, II. Template functions and composition as determined by transcription with RNA polymerase. Proc. Natl. Acad. Sci. USA 60, 915-920.
KLECKNER, N. & WEINER, B. M. (1993). Potential advantages of unstable interactions for pairing of chromosomes in meiotic, somatic and premeiotic cells. Cold Spring Harbor Symp. Quant. Biol. 58, 553-565.
LIEBOVITCH, L. S., TAO, Y., TODOROV, A. T. & LEVINE, L. (1996). Is there an error-correcting code in the base sequence of DNA? Biophys. J. 71, 1539-1544.
LOBRY, J. R. (1995). Properties of a general model of DNA evolution under no- strand-bias conditions. J. Mol. Evol. 40, 326-330.
LOBRY, J. R. (1996a). Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 13, 660-665.
LOBRY, J. R. (1996b). Origin of replication of Mycoplasma genitalium. Science 272, 745-746.
MCGEOCH, D. J., DALRYMPLE, M. A., DAVISON, A. J., DOLAN, A., FRAME, M. C., MCNAB, D., PERRY, L. J., SCOTT, J. E. & TAYLOR, P. (1988). The complete DNA sequence of the long unique region in the genome of Herpes simplex virus type 1. J. Gen. Virol. 69, 1531-1574.
MCKEE, B. D. (1996). Meiotic recombination: a mechanism for tracking and eliminating mutation. BioEssays 18, 411-419.
MURCHIE, A. I. H., BOWATER, R., ABOUL-ELA, F. & LILLEY, D. M. J. (1992). Helix opening transitions in supercoiled DNA. Biochem. Biophys. Acta 1131, 1-15.
NUSSINOV, R. (1981). Eukaryotic dinucleotide preference rules and their implications for degenerate codon usage. J. Mol. Biol. 149, 125-131.
OLIVER, S. G. et al., (1992). The complete DNA sequence of yeast chromosome III. Nature 357, 38-46.
PRABHU, V. V. (1993). Symmetry observations in long nucleotide sequences. Nucleic Acids Res. 21, 2797-2800. (Click Here)
ROCCO, V. & NICOLAS, A. (1996). Sensing of DNA non-homology lowers the initiation of meiotic recombination in yeast. Genes to Cells 1, 645-661.
SHEMER, R., BIRGER, Y., DEAN, W. L., REIK, W., RIGGS, A. D. & RAZIN, A. (1996). Dynamic methylation adjustment and counting as part of imprinting mechanisms. Proc. Natl. Acad. Sci. USA 93, 6371-6376.
SIBBALD, P. R., BANERJEE, S. & MAZE, J. (1989). Calculating higher order DNA sequence information measures. J. theor. Biol. 136, 475-483.
SINGER, M. J. & SELKER, E. U. (1995). Genetic and epigenetic inactivation of repetitive sequences in Neurospora crassa. RIP, DNA methylation and quelling. Curr. Top. Microbiol. Immun. 197, 165-177.
SMITH, J. M. (1989). Evolutionary Genetics. pp. 257-270, Oxford, UK: Oxford University Press.
SMITHIES, O., ENGELS, W. R., DEVEREUX, J. R., SLIGHTOM, J. L. & SHEN, S. (1981). Base substitutions, length differences and DNA strand asymmetries in the human Gl and A l fetal globin gene region. Cell 26, 345-353.(Click Here)
SUEOKA, N. (1995). Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 40, 318-325.
TOMIZAWA, J. (1984). Control of ColE1 plasmid replication: the process of binding of RNA I to the primer transcript. Cell 38, 861-870.
TRACY, R. B., CHEDIN, F. & KOWALCZYKOWSKI, S. C. (1997). The recombination hot-spot Chi is embedded within islands of preferred DNA pairing sequences in the E. coli genome. Cell 90, 205-206.
TURNER, D. H. (1996). Thermodynamics of base pairing. Curr. Opin. Struct. Biol. 6, 299-304.
WATSON, J. D. & CRICK, F. H. C. (1953). Genetical implications of the structure of deoxyribonucleic acid. Nature 171, 964-967.
YURA, T., MORI, H., NAGAI, H., NAGATA, T., ISHIHAMA, A., FUJITA, N., ISONO, K., MIZOBUCHI, K. & NAKATA, A. (1992). Systematic sequencing of the Escherichia coli genome: analysis of the 0-2.4 min region. Nucleic Acids Res. 20, 3305-3308.
| End
Note 2002:
The following five quotations are taken from a paper entitled "Compositional Symmetries in Complete Genomes" by Dong Qi and A. Jamie Cuticchia of the Bioinformatics Supercomputing Centre, The Hospital for Sick Children, Toronto. The paper was published in 2001 in Bioinformatics 17, 557-559. Using the "SEARCH" facility on the HomePage of this site, or the "FIND" facility of your web browser, you can determine that the same sentences appear in the above paper (Bell and Forsdyke, 1999a).
|
The possibility of this having occurred by chance being virtually impossible, this fits the formal definition of plagiarism. The copyright agreement Dr. Cuticchia would have had to sign for the paper to be published by Oxford University Press reads:
However, plagiarism may not be deliberate. Some senior scientists who have busy schedules rely on their associates to write papers. The latter, whose first language may not be English, may at one point in time take direct quotations to their notes. At a later point in time when asked to put together a paper, they may forget that the notes were in the form of direct quotations. If the senior colleague then puts his own name on the paper he accepts full responsibility for what has been written but, with respect to the direct quotations, he is guilty only of professional negligence, not of deliberate plagiarism. The use of the direct quotations would seem to establish that the original paper (Bell and Forsdyke, 1999a), although not cited, must have been read very carefully. The author could not have been unaware of Prabhu's 1993 paper entitled "Symmetry observations in long nucleotide sequences." The title of Qi and Cuticchias' 2001 paper was "Compositional symmetries in complete genomes." It is regrettable that there was no citation of Prabhu's 1993 paper, since, as pointed out by Forsdyke (2002) in a "commentary" on the Qi-Cuticchia paper (Bioinformatics 18, 215-217), there was a great deal of overlap between the two papers (Click Here). There was, however, citation of other works (e.g. Bell and Forsdyke, 1999b). In that the two papers of Bell and Forsdyke were published together in the same journal issue, the author of the Qi-Cuticchia paper might have presumed that a reader of one (1999b) would have seen the other (1999a), and so could become aware of Prabhu's 1993 paper. Authors are often under pressure from publishers to decrease the number of references, and this is one way of complying. However, the opening paragraph of the Qi-Cuticchia paper boldly declares:
Perhaps the author(s) of a paper claiming to "reveal a universal parity rule," should have exercised more care. Unfortunately, at the time the paper appeared Dr. Cuticchia was much engaged in one of the many scandals which plague The Hospital for Sick Children (see Nature 2001, 414, 384, and Toronto Globe and Mail, 12th and 15th Nov. 2001; (Click Here)). |
| End Note 2005. The originator in 1909 of the word "gene" W. L. Johannsen thought of genes as accounting units (Rechnungseinheiten). |
Go to: Deviations from Chargaff's Second Parity Rule (Bell & Forsdyke 1999b) (Click Here)
Go to: Prabhu 1993 (Click Here)
Return to: Bioinformatics (Genomics) (Click Here)
Return to: HomePage (Click Here)
For: Cryptology and the Rosetta Stone (Click Here)
Last edited 26 Apr 2011 by Donald Forsdyke