Introns First
By Donald R. Forsdyke
Final pub
Received 3 June 2012 - Accepted 22 Jan 2013
Published online 9 Feb 2013 by Springer for The Konrad Lorenz Institute for Evolution and Cognition Research (copyright holder)
![]()
Negative_Role_of_Recombination
Exons_as_Another_Line_of_Defence
Exons_and_Introns_Defend_by_Changing_GC%
GC%_Differences_Affect_Stem-Loop_Extrusion
End_Note_(Feb_2013) Werner Callebaut (1952-2014)
End_Note_(Nov_2014) Are introns a burden?
![]()
|
|
This
paper was presented at
"Genome Brno 2," a workshop on "Structural and Functional Diversity of
Genomes" held September 2012 at
the Augustinian Abbey of St. Thomas, Brno, Czech
Republic. The You Tube internet site hosts the talk, and also
available is an extended, more elementary, version - "Introns and Exons"
- as an
18 video course for high school and college students (Click Here). |
Sometimes it is
important to know the order of events. But sometimes this seems an academic
exercise. Did giraffes with longer necks better detect approaching predators and
then, as a useful by-product, become less vertically challenged in their
grazing? Or could it have been the other way round? And, in the unlikely event
that we could obtain a definite answer, would it really matter (Wilkinson and
Ruxton 2012)? For introns and exons it would matter. In the
likely event that we
will obtain a definite answer to
whether, in the general case, one preceded the other, I show here that we will
have greatly enhanced our understanding of the information we carry in our DNA.
For the general case, the alternatives are easily set out: Segments of DNA with
properties that we would now deem as intronic, and segments of DNA with
properties that we would now deem exonic, arose simultaneously. On the other
hand, exons might have preceded introns, or introns might have preceded exons.
And if exons were first, did introns arise early, or late, in evolutionary time?
Likewise, if introns were first, did exons arise early, or late?
Before these questions could be asked, exons and introns had
to be discovered. Prior to this, genomes had been neatly divided into genic
(usually protein-encoding) and non-genic sectors. Then in 1977 the unexpected
discovery that genes were "split" or "interrupted" by non-protein-encoding
segments, led to the coinings "exon" and "intron," and a flurry of speculation
as to intron origins (Gilbert 1978; Reanney 1978,1979; Darnell 1978; Crick
1979). It was easy to think of introns as "informationally irrelevant"
(Doolittle 1978), an idea consistent with the view that our genomes were laden
with "junk DNA," perhaps with devilishly selfish intent (Orgel, Crick and
Sapienza 1980). As facts and arguments contended with counterfacts and
counterarguments, the fortunes of various hypotheses waxed and waned over
succeeding decades. However, some in the field recently pronounced that at last
"a degree of clarity has been reached in the study of the evolution of
eukaryotic gene architecture". While not attempting "a comprehensive coverage,"
yet dealing with "several aspects that appear directly relevant for
understanding evolution of introns and eukaryote gene structure," they concluded
that "an introns first scenario is not supported by any evidence" (Rogozin et al.
2012). I agree that some clarity has been reached, but it is a clarity that
allows us merely to begin sorting out the various hypotheses in the light of the
accumulated evidence. I will argue here that at least one version of "introns
first," which is featured in two textbooks (Forsdyke 2011a, 2011b), is still
very much in contention.
Introns emerged
with the systematic application of new technology. Just as the phenomenon now
known as genetic linkage emerged around 1900 when Bateson and Saunders applied
Mendelian technology (brother-sister matings through the generations) and found
certain characters to be "partially coupled," or "coupled," rather than
independently inherited (Cock and Forsdyke 2008), so the intron phenomenon
emerged in 1977 when electron microscopy was applied to the visualization of
R-loops in hybridized DNA molecules (Witkowski 1988; Morange 1998). This was
soon supported by another new technology - DNA sequencing (Gilbert 1981). Here
we are concerned with yet another new technology, the computer calculation of
optimum secondary structure in single-stranded nucleic acid sequences, which can
be represented both pictorially and as a stability number (expressed in negative
kilocalories per mol.; Le and Maizel 1989; Zuker 1990). For such structures
there are three determinants - the types and order of bases, and sequence
length. If, for purposes of comparison, lengths are kept constant, then there
are just two determinants, base composition and
order. These can be considered
independently as base composition-dependent and base order-dependent
stabilities, which sum together to give the total stability (Forsdyke 2007a).
The base order-dependent component can be determined by subtracting the base
composition-dependent component from the total stability value (Fig. 1). Before
moving to the application of this technology, I first review various ideas that
followed the discovery of introns.
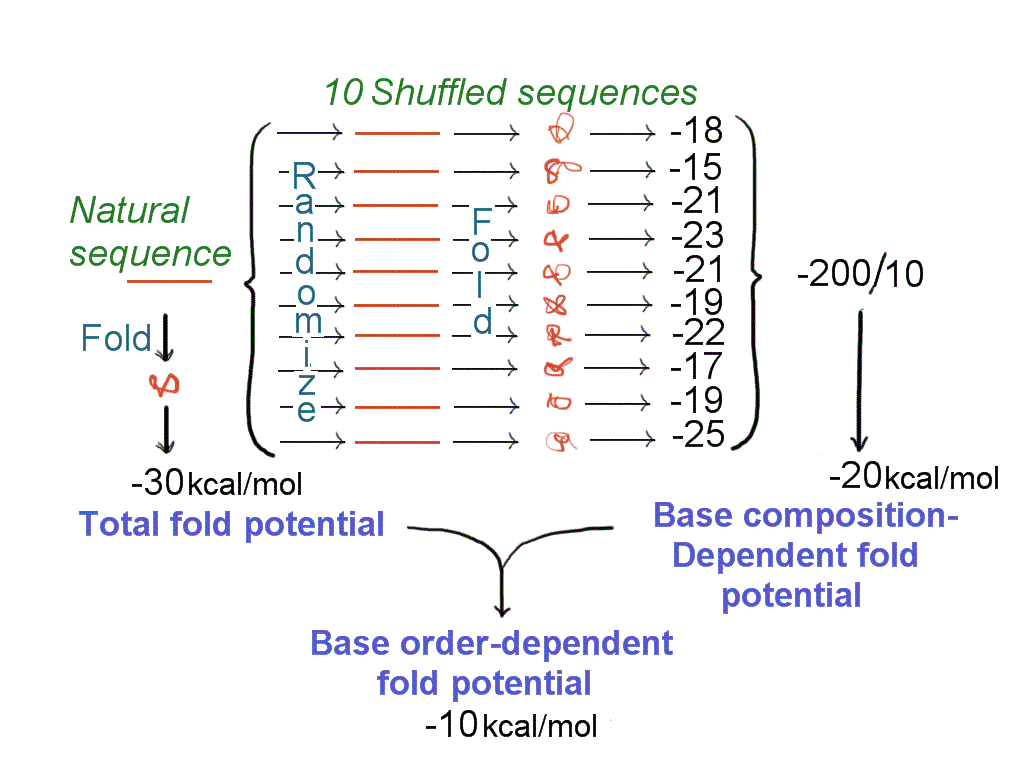 |
|
Fig 1
Determination of base order-dependent
fold potential. A natural sequence (horizontal line at left) is computer folded
by successive reiteration to obtain a structure of stability -30 kcal/mol (total
fold potential). Base order is randomized to produce ten shuffled sequences that
share with the natural sequence only their base composition. These are each
folded to obtain corresponding stability values. Idiosyncracies due to each
distinctive base order are averaged out (at right) to determine the contribution
of base composition (-20 kcal/mol) to the total fold potential of the natural
sequence. The contribution of base order to the total fold potential of the
natural sequence is determined by subtraction |
Positive Role in
Recombination
Early in 1978,
within a few months of their discovery, one of the sequencing pioneers, Walter
Gilbert, suggested that introns had arisen to facilitate recombination (Gilbert
1978). As had been pointed out by Thomas Hunt Morgan (1911), the more closely
coupled are two genes, the less likely will their coupling be broken by
recombinational shuffling. Gilbert suggested that if the length of an individual
gene were increased by virtue of its introns, the probability of recombination
between different parts of that gene would be increased. By creating new
combinations of parts of the gene product (e.g. the shuffling of protein
domains), the number of variant forms offered to natural selection would be
increased. This could speed up evolution if variation, not selection for that
variation, was rate-limiting.
suggested that introns had arisen to facilitate recombination (Gilbert
1978). As had been pointed out by Thomas Hunt Morgan (1911), the more closely
coupled are two genes, the less likely will their coupling be broken by
recombinational shuffling. Gilbert suggested that if the length of an individual
gene were increased by virtue of its introns, the probability of recombination
between different parts of that gene would be increased. By creating new
combinations of parts of the gene product (e.g. the shuffling of protein
domains), the number of variant forms offered to natural selection would be
increased. This could speed up evolution if variation, not selection for that
variation, was rate-limiting.
Gilbert went further to suggest that an organism that
modified the sequence of its introns to favor recombination would be at a
selective advantage: "Middle repetitious sequences within introns may create hot
spots for recombination to rearrange the exonic sequences." At that time a view
similar to Gilbert's, involving conservation of intron sequences with the
potential to form stem-loop structures that would engage in "topological
reshufflings," was advanced by Darryl Reanney (1978). But, noting the high
mutation rate in the intron sequences then available, Gilbert soon shifted
position declaring that "it is not their sequence that is relevant, but their
length. Their function is to move the exons apart along the chromosome" (Gilbert
1981). Some, however, doubted Nature's prescience in creating introns in the
hope of opportunities for recombination many generations later. It seemed
necessary that introns should have offered some more proximate advantage
(Doolittle 1978; Crick 1979).
Negative Role of
Recombination
The seemingly high mutation rate in introns prompted Philip
Leder and his colleagues in 1978 to
 propose that introns would speed up
evolution, not by enhancing, but by
preventing recombination (Tiemeier et al. 1978). Supposing an adaptive
advantage for the generation of globin gene duplicates within an organism, they
noted that: "Immediately after the original duplication event, it is probable
that extensive homology existed between these gene segments." Since
recombination required close sequence identity, variation in introns "would
reduce the target size for possible recombination and serve to stabilize or fix
the two - - globin gene copies." Noting that the "free and easy nucleotide
substitution that occurs in introns should serve as a buffer against
mispairing," a respected arbiter, Russell Doolittle (1985), came to agree that,
whether introns supported recombination and "exon shuffling" as proposed by
Gilbert, or decreased recombination as proposed by Leder, it would be much to
the advantage of an organism to have them. However, the question as to whether
one role or the other (or neither) had been instrumental in the actual origin of
introns, was left unanswered.
propose that introns would speed up
evolution, not by enhancing, but by
preventing recombination (Tiemeier et al. 1978). Supposing an adaptive
advantage for the generation of globin gene duplicates within an organism, they
noted that: "Immediately after the original duplication event, it is probable
that extensive homology existed between these gene segments." Since
recombination required close sequence identity, variation in introns "would
reduce the target size for possible recombination and serve to stabilize or fix
the two - - globin gene copies." Noting that the "free and easy nucleotide
substitution that occurs in introns should serve as a buffer against
mispairing," a respected arbiter, Russell Doolittle (1985), came to agree that,
whether introns supported recombination and "exon shuffling" as proposed by
Gilbert, or decreased recombination as proposed by Leder, it would be much to
the advantage of an organism to have them. However, the question as to whether
one role or the other (or neither) had been instrumental in the actual origin of
introns, was left unanswered.
There was much excitement when the
various domains of immunoglobulins seemed in accord with the Gilbert hypothesis,
introns being located at domain boundaries (Robertson 1977). But as more protein
sequences were examined, immunoglobulins came to be viewed as special cases
(Crick 1979). And even though Gilbert (1978) had drawn attention to the fact
that "genes with no protein product, such as the tRNA genes in yeast and the
rRNA genes in Drosophila," contained
introns, at first it was easy also to dismiss non-protein-encoding genes as
special cases, despite the finding that the 5' and 3' non-coding regions of some
protein-encoding genes contained introns (Crick 1979). When, in the 1990s, long
non-coding RNAs laden with introns became evident (Pfeifer and Tilghman 1994),
such dismissal seemed less valid. Those who had been trying to relate intron
positions to boundaries between protein domains had been following a false
trail. And the notion of introns as places for "free and easy nucleotide
substitution" was also loosing ground. Walter Schaffner and his colleagues
pointed to the "long known paradox - - that most introns are preserved even though
their actual sequence hardly seems to matter" (Matsuo et al. 1994).
Exons as Another Line
of Defence
Supporting
Leder's case for a negative role of introns in recombination, Schaffner noted
that "even the few sequence mismatches in introns that typically occur between
different strains can dramatically lower
the efficiency of homologous recombination" (my italics). Thus, in agreement
with Leder's "homology interruption hypothesis," he proposed, from studies of
the POU domain transcription factor gene family, that there would be a "homology-reducing effect of divergent introns". But Schaffner then went much
further (Matsuo et al. 1994). If it was important to decrease recombination, why
leave it to introns? Exons might help: "The frequency of homologous
recombination among POU domain genes could be reduced not only by interrupting
exons with introns, but also by minimizing sequence identity within exons." And
would this exon identity-minimization be random, or would some base changes be
more effective than others? While introns might represent "a first barrier"
against homologous recombination between members of gene families, another "line
of defense" would be "the overall sequence composition and especially
synonymous codon choice" [my italics].
Exons and Introns Defend by Changing GC%
On checking
actual sequences, the Schaffner group found that, while the amino acid sequences
of the various POU domains (in genes encoding Oct-1, Oct-2, and Pit-1) tended to
remain identical, the corresponding exon sequences were "quite different." This
meant that, rather than changes in bases essential for specifying amino acids
(first and second codon positions), it was changes in the remaining sequence
(third codon positions) that would lower recombination efficiency. They observed
that: "The G+C content of the Oct-2 POU domain DNA is high, while that of the
Oct-1 POU domain is intermediate," and "the Pit-1 POU domain is A+T-rich." Thus
they related failure of these genes to recombine with each other, to switches
between synonymous codons that would change the GC% (i.e. changes at third
positions). Since the GC% values of synonymous codon positions resembled that of
introns (D'Onofrio et al. 1991; Vinogradov 2001), then failure to recombine
would also associate with intronic GC% values. In other words, both the first
and second "lines of defence" might be using the
same weapon against recombination -
differences in GC%. By the same token, similarities in GC% might favor
recombination. But how GC% values might affect recombination remained to be
explained.
As usually
employed, the terms "introns early" and "introns late" refer to whether exons
acquired introns early, or late, in evolution, with the implication that exons
arose simultaneously with, or preceded, introns. Although it can be considered a
subset of "introns early," the term "introns first" implies that segments of DNA
with properties that we would now deem intronic
preceded exons. In other words, nucleic acid sequences were to some
degree, and perhaps entirely, intronic in nature, and segments became actually
defined as intronic when they acquired exon borders (Penny et al. 2009).
The "introns first" idea was advanced by Darryl Reanney
(1979). He portrayed the RNA splicing required to remove intron segments from
primary RNA transcripts as a hold-over from early life forms in an "RNA world."
This preceded the evolution of modern forms where the DNA 'legislature'
(information source) is largely dissociated from the protein 'executive,' which
is specified by that information. In the RNA world there were no proteins, and
RNA molecules were their own legislature and executive. Indeed, Reanney saw that
the "'mosaic' RNAs produced by splicing are
strictu sensu recombinant molecules in
that they contain data drawn from different parts of the genome. - - It seems
logical therefore to suggest that RNA:RNA splicing is the primitive mode of
genetic recombination." Pointing to an important role of "previous folding" in
interactions between separate single-stranded RNAs, Reanney deduced that
"topology must have had a key role in the selection of the splice site - -
explicable if nucleotides near the site are required to be unpaired in order to
provide a recognition mechanism through base pairing with an independent RNA."
At that time
agreeing with Gilbert on the adaptive advantages of segment shuffling (i.e. a
function that increased variation),
Reanney went further to suggest that "this type of recombination developed from
the proofreading function which seems to be a universal correlate of DNA
synthesis" (i.e. a function that decreased
variation). Thus today's "generalized recombination could be regarded as an
extension of the
 proofreading function from which it evolved." However, as the
"introns early" case (Darnell 1978, Doolittle 1978) grew stronger, Reanney
(1984) took a firmer position on the importance of
decreasing variation to the origin of introns.
proofreading function from which it evolved." However, as the
"introns early" case (Darnell 1978, Doolittle 1978) grew stronger, Reanney
(1984) took a firmer position on the importance of
decreasing variation to the origin of introns.
If proof-reading was so necessary in the present DNA world,
then it might have been even more necessary in the earlier RNA world. In this
world all "genes" would have been part of error-prone RNA molecules and their
evolution would have depended on the parallel development of mechanisms for
detecting and correcting errors. To this extent, it could be said that "genes"
and introns arose hand-in-hand. But, in the context of the later-appearing
protein-encoding genes, the scenario can be better described as "introns first"
(Penny et al. 2009). It seems likely that prior development of sophisticated
error detection and correction capacities would have been critical for genomes
to evolve protein-encoding capacity.
Positive Role in Error-Detection
Noting that the error-free transmission of electronic information requires the interruption of message sequences by non-message, error-detecting, sequences, which operate by parity-check mechanisms (Hamming 1980), I suggested a parity check mechanism for the operation of error-detecting sequences in introns (Forsdyke 1981). Although gaining no clear supported from subsequent studies (Liebovitch et al. 1996; Battail 2007; Faria et al. 2012), four key postulates still seem valid:
-
1. Introns contribute positively to the hereditary transmission of error-free genetic information.
-
2. The structure of single-stranded DNA is involved.
-
3. Some form of parity between bases is involved.
-
4. The error-detecting function affects DNA pervasively at regular intervals - just as breathing interrupts human speech, or as adverts ("pauses for station identification") interrupt TV programs.
These postulates provided the basis for a recombination-dependent
error-checking mechanism, the conception of which began with the unearthing of
Chargaff's, long-forgotten, second parity rule.
Chargaff's
first parity rule provided a basis for the Watson-Crick structure for duplex
DNA, namely
 that
there was parity between purines on one strand and pyrimidines in the other - base A pairing with base T, and base G pairing with base C
(Chargaff 1951). By the same token, the dinucleotide AG on one strand would pair
with the dinucleotide CT on the antiparallel complementary strand - a rule that
applied to all 16 possible dinucleotides. Similarly, trinucleotide AGT would
pair with complementary trinucleotide ACT on the opposite strand of a duplex,
and this would apply for all 32 pairs of possible trinucleotides. This numerical
parity between oligonucleotides on complementary strands followed naturally from
the structure of duplex DNA.
that
there was parity between purines on one strand and pyrimidines in the other - base A pairing with base T, and base G pairing with base C
(Chargaff 1951). By the same token, the dinucleotide AG on one strand would pair
with the dinucleotide CT on the antiparallel complementary strand - a rule that
applied to all 16 possible dinucleotides. Similarly, trinucleotide AGT would
pair with complementary trinucleotide ACT on the opposite strand of a duplex,
and this would apply for all 32 pairs of possible trinucleotides. This numerical
parity between oligonucleotides on complementary strands followed naturally from
the structure of duplex DNA.
Chargaff's second parity rule was that the first parity rule
equivalences also apply pervasively, although not quite so precisely, to DNA
single strands (Rudner et al. 1968). As with the first rule, the equivalences
also extended to oligonucleotides (Prabhu 1993). An implication of this was
that, single stranded DNA, although normally forming part of a duplex, could
have an independent potential to form stem-loop structures that, at least in the
stems, would exhibit parity between complementary bases and oligonucleotides
(Fig. 2).
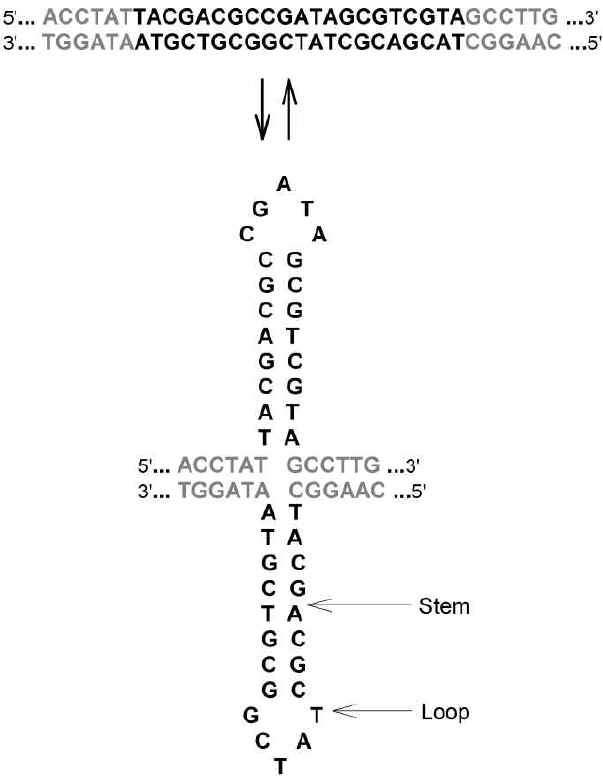 |
|
Fig 2
The potential of a DNA duplex (top) to
reversibly extrude stem-loops (bottom) when complementary oligonucleotide sequences are
both present in equal quantities on one strand (Chargaff's second parity rule),
and closely located. Here the octonucleotide TACGACGC in the top strand,
complements the octonucleotide GCGTCGTA in the same strand, to form the stem in
an extruded secondary structure. A symmetrical stem-loop is formed by
corresponding octonucleotides in the bottom strand |
A new technology - computer-aided structure determination (Fig. 1) - facilitated the demonstration that the potential for the extrusion of stem-loop structures from duplex DNA ("fold potential") was pervasively distributed along the DNA molecules of numerous biological species (Forsdyke 1995a-c; 1996). Furthermore, when decomposed into base order-dependent and base composition-dependent components, it was evident that fluctuations in fold potential were largely due to the base order-dependent component (Zhang et al. 2008a; Fig. 3). The latter provided a powerful means of analyzing the distribution of fold potential between introns and exons.
 |
|
Fig 3
Potential stabilities of secondary
structures extruded from duplex DNA, as assessed for a 200 base window moved in
50 base steps along the sequence. Total stability values (A) decompose into
base
order-dependent components (B) and base composition-dependent components (C).
The
distribution of values for the top strand (blue line) and bottom strand (red
line) closely correspond. This is a 40 kb segment of chromosome I of C.
elegans (nucleotides 2500 to
42500) |
That fold potential was much greater in introns was particularly evident in the case of genes under positive Darwinian selection, where introns could be more conserved than exons (Fig. 4); but high intronic fold potential could also be demonstrated in other genes (Forsdyke 1996; Dawson and Yamamoto 1999; Bechtel et al. 2008).
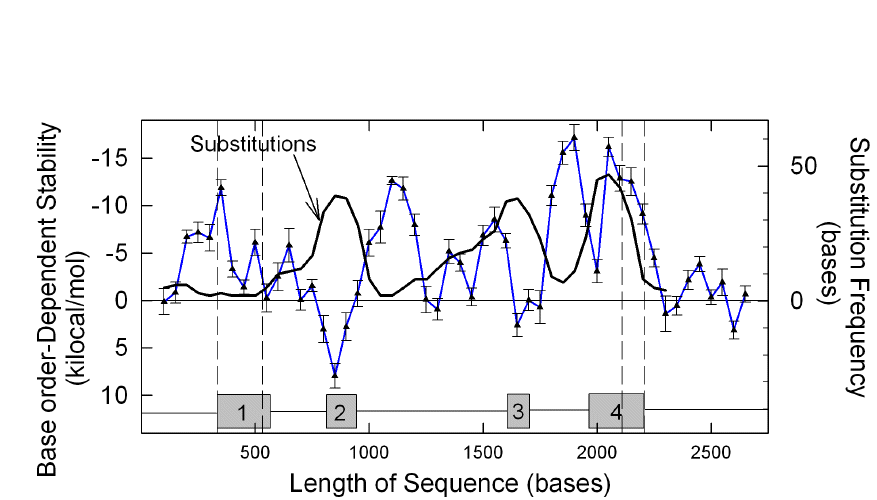 |
|
Fig 4
In a gene under positive Darwinian
selection, conserved introns harbor enhanced stem-loop potential, assessed as
stability of secondary structure. Conversely, there is high
base substitution
frequency (continuous black line) and low base order-dependent fold potential
(blue line with triangles) in exons. The numbered grey boxes indicate the
locations of the four exons of the rattlesnake venom gene encoding a basic
subunit of venom phospholipase A2; dashed vertical lines show,
consecutively, the beginning of exon 1, the beginning of the protein-coding part
of exon 1, the end of the protein-coding part of exon 4, and the end of exon 4.
Values were determined for a 200 base window moving in 50 base steps along the
sequence. Substitutions are base differences relative to the rattlesnake
phospholipase A2 acidic subunit gene. The two genes are likely to
have arisen from a common ancestral gene. A similar result was obtained when the
comparison was with Habu snake phospholipase A2 |
The greater ability of introns to order bases to support the extrusion of
stem-loop structures from duplex DNA was readily rationalized in terms of
conflicting pressures. In exons the pressure to order bases for stem-loop
potential ("fold pressure") would conflict with the pressure to encode amino
acids ("protein pressure"). Third codon position, being less subject to protein
pressure, could be seen as "mini-introns" that would allow some base ordering to
support fold potential in exons.
To initiate
legitimate recombination - homologous recombination - between nucleic acids,
there must
 be pairing between complementary, or closely complementary,
sequences. Crick (1971) proposed that, for this to occur between two duplex DNA
molecules, the strands in each duplex would need to unpair locally so that they
could test each other for complementarity. But the finding of Jun-ichi Tomizawa
that recombining complementary single-stranded RNA molecules first interact by
reversible "kissing" between the loops of stem-loop structures (Tomizawa 1984),
prompted Kleckner and Weiner (1993) to suggest that the locally unpaired single
stranded DNA molecules would also interact by way of loop-loop interactions.
Since recombination was a genome-wide characteristic of DNA, this was consistent
with the observed pervasive distribution of stem-loop potential throughout
genomes. This pervasive pressure would account for the need for protein-encoding
capacity to arise in segments (exons) interrupted by segments with high
stem-loop potential. Much evidence supporting the role of stem-loops in
generalized recombination has since accumulated (Forsdyke 2007b). But how would
recombination allow error-detection and correction?
be pairing between complementary, or closely complementary,
sequences. Crick (1971) proposed that, for this to occur between two duplex DNA
molecules, the strands in each duplex would need to unpair locally so that they
could test each other for complementarity. But the finding of Jun-ichi Tomizawa
that recombining complementary single-stranded RNA molecules first interact by
reversible "kissing" between the loops of stem-loop structures (Tomizawa 1984),
prompted Kleckner and Weiner (1993) to suggest that the locally unpaired single
stranded DNA molecules would also interact by way of loop-loop interactions.
Since recombination was a genome-wide characteristic of DNA, this was consistent
with the observed pervasive distribution of stem-loop potential throughout
genomes. This pervasive pressure would account for the need for protein-encoding
capacity to arise in segments (exons) interrupted by segments with high
stem-loop potential. Much evidence supporting the role of stem-loops in
generalized recombination has since accumulated (Forsdyke 2007b). But how would
recombination allow error-detection and correction?
Recombination and Error-Correction
Reanney (1979)
stated:
|
Here Reanney is describing
error-detection and correction by the process which, when it occurs in a genic
region, is referred to as gene conversion - the directional transfer of
information from a gene on one chromosome to that on another (Yang et al. 2012).
What Reanney called "preferential correction to wildtype," was a topic of my
first introns paper (Forsdyke 1981). If there is an error in a text, you want
the error to be noted and corrected, not compounded. In other words, if you are
comparing two lines of text (or two strands of DNA) you want to know, not
merely that there has been an error, but which line is the wrong line and which
line is the correct line. Information in the latter is used to correct the
former.
Reanney
pointed to strand marking by methylation. Instead of erasure, some methyl marks
can persist transgenerationally. So DNA, in computer jargon, is not just
"read-only memory", but "read-and-write memory," with the writing persisting for
at least a few generations. A modification of this epigenetic marking was
suggested by Virgil Reese (2002). He noted that the cell either "knows" which
strand is incorrect, or is uncertain. In the latter case, it can mark strands as
"suspicious" by methylation. Sometimes the methyl mark can be carried through to
future generations where the suspiciously marked strand may find itself paired
with a non-suspiciously marked strand. Correction from the latter can then be
implemented.
The trouble with this, as noted with some intron hypotheses, is that Nature has
to do something with no adaptive advantage in one generation in the hope that it
will be useful to a future generation. Does Nature have such foresight?
Sometimes something useful in one generation can be adapted for another role in
a future generation. In the nineteenth century Samuel Butler (1926) noted: "I
have gone out sketching and forgotten my water-dipper; among my traps I always
find something that will do, for example, the top of my tin case (for holding
pencils). This is how organs come to change their uses and hence their forms, or
at any rate partly how." Today we make the same point
with the "spandrels" metaphor (Gould 1993). Once introns were in existence,
there was indeed ample opportunity for them to assume other roles, such as
domain shuffling (Gilbert 1978), harboring regulatory and "selfish" elements
(Orgel et al. 1980), preventing recombination (Tiemeier et al. 1978) and
developing certain asymmetries between top and bottom strands that violate
Chargaff's second parity rule (Forsdyke and Bell 2004; Zhang et al. 2008b). We
can also note that methylation predisposes a strand to exchange a T residue for
a C residue. Thus, an initial transient epigenetic event has the potential to
influence our genomes more permanently. The "writing" became indelible.
GC% Differences Affect Stem-Loop Extrusion
The ability of
introns to defend against recombination, was seen by the Leder and Schaffner
groups as a way of preserving paralogous genes within members of a species. For
this Schaffner pointed to a role of differences in base composition - GC% - but
left the mechanism unexplained. Along the lines of Le and Maizel (1989), it has
been shown that the structure of extruded stem-loops would be sensitive to very
small differences in GC% (Forsdyke 2007b, 2011c). This should suffice to prevent
recombination. By the same token, it has been argued that base composition
differences between two members of a species would serve to prevent the meiotic
pairing of their chromosomes in the gonads of their offspring, so enforcing
their reproductive isolation - an isolation that could lead to branching
speciation (Fig. 5). Thus, seeking to know how introns originate, today helps us
approach
 |
|
Fig 5
Similar extruded stem-loop structures
when GC percentage values are equal (left), favor strand pairing, and hence,
favor recombination. Dissimilar extruded stem-loop structures when GC percentage
values are not equal (right), prevent strand pairing, and hence, prevent
recombination. The potentially homologous DNA duplexes, of either paternal (P)
or maternal (M) origin, would attempt to pair during meiosis in the gonad of
their child (hybrid). The hybrid is either fertile (left) or sterile (right) |
Acknowledgement
Queen's
University hosts my intron webpages
Click Here.
References
Battail G (2007) Information theory and
error-correcting codes in genetics and biological evolution. In: Barbieri M (ed)
Introduction to biosemiotics. The new biological synthesis.
Springer, Dordrecht,
pp 299-345
Bechtel JM, Wittenschlaeger T, Dwyer T, Song J,
Arunachalam S, Ramakrishnan SK, Shepard S, Fedorov A (2008) Genomic mid-range
inhomogeneity correlates with an abundance of RNA secondary structures.
BMC
Genomics 9:284 doi:10.1186/1471-2164-9-284
Butler S (1926)
The Shrewsbury edition of the works of Samuel
Butler. Jones HF, Bartholomew AT (eds) Vol. 20,
Jonathan Cape, London, p. 13
Chargaff
E (1951) Structure and function of nucleic acids as cell constituents.
Fed Proc 10:654-659
Cock AG, Forsdyke DR (2008)
Treasure your exceptions.
The science and life of William Bateson. Springer, New York, pp 339-377
Crick F (1971) General model for chromosomes of higher
organisms. Nature 234:25-27
Crick F (1979) Split genes and RNA splicing.
Science
204:264-271
Darnell JE (1978) Implications of RNA-RNA splicing in
evolution of eukaryotic cells. Science 202:1257-1260
Dawson WK, Yamamoto K. (1999) Mean free energy topology
for nucleotide sequences of varying composition on secondary structure
calculations. J Theor Biol 201:113-140
D'Onofrio G, Mouchiroud D, A�ssani B, Gauter C,
Bernardi G (1991) Correlations between the compositional properties of human
genes, codon usage, and amino acid composition of proteins.
J Mol Evol
32:504-510
Doolittle RF (1985) The genealogy of some recently
evolved vertebrate proteins. Trends Biochem Sci 10:233-237
Doolittle WF (1978) Genes in pieces: were they ever
together? Nature 272:581-582
Faria LCB, Rocha ASL, Kleinschmidt JH, Silva-Filho MC,
Bim E, Herai RH, Yamagishi MEB, Palazzo R (2012) Is a genome a codeword of an
error-correcting code? PLOS One 7 (5) e36644. Doi:10.1371/journal.pone.0036644
Forsdyke DR (1981)
Are
introns in-series error detecting sequences?
J Theor Biol 93:861-866
Forsdyke DR (1995a) A stem-loop "kissing" model for the
initiation of recombination and the origin of introns.
Mol Biol Evol
12:949-958
Forsdyke DR (1995b) Conservation of
stem-loop potential in introns of snake venom phospholipase A2 genes:
an application of FORS-D analysis.
Mol Biol Evol
12:1157-1165
Forsdyke DR (1995c) Relative roles of primary sequence
and (G+C)% in determining the hierarchy of frequencies of complementary
trinucleotide pairs in DNAs of different species. J Mol Evol
41:573-581
Forsdyke DR (1996) Different biological species
"broadcast" their DNAs at different (G+C)% "wavelengths".
J Theor Biol
178:405-417
Forsdyke
DR (2007a) Calculation of folding energies of single-stranded nucleic acid
sequences: conceptual issues.
J Theor Biol
248:745-753
Forsdyke DR (2007b) Molecular sex: the importance of
base composition rather than homology when nucleic acids hybridize.
J Theor
Biol 249:325-330
Forsdyke DR (2011a) The interrupted gene. In:
Lewin's
genes X. Krebs JE, Goldstein ES, Kilpatrick ST (eds) Jones and Bartlett, Boston,
pp. 79-97, 172-175
Forsdyke DR (2011b)
Evolutionary bioinformatics, 2nd
edition. Springer,
Forsdyke DR (2011c) The selfish gene revisited:
reconciliation of Williams-Dawkins and conventional definitions.
Biol Theor
5:246-255
Gilbert W (1978) Why genes in pieces?
Science 271:501
Gilbert W (1981) DNA sequencing and gene structure.
Science 214:1305-1312
Gould SJ (1993) Fulfilling the spandrels of world and
mind. In:
Understanding scientific prose. Selzer J (ed) University of Wisconsin
Press, Madison, pp. 310-336
Hamming RW (1980)
Coding and information theory.
Prentice-Hall,
Kleckner N, Weiner BM (1993) Potential advantages of
unstable interactions for pairing of chromosomes in meiotic, somatic and
premeiotic cells.
Cold Spring Harb Symp Quant Biol
58:553-565
Le S-Y, Maizel JV (1989) A method for assessing the
statistical significance of RNA folding.
J Theor Biol
138:495-510
Liebovitch LS, Tao Y, Todorov AT, Levine L (1996) Is
there an error-correcting code in the base sequence of DNA?
Biophys J
71:1539-1544
Matsuo K, Clay O, Kunzler P, Georgiev O, Urbanek P,
Schaffner W (1994) Short introns interrupting the Oct-2 POU domain may prevent
recombination between POU family genes without interfering with potential POU
domain 'shuffling' in evolution. Biol Chem Hoppe-Seyler
375:675-683
Morange M (1998) A history of molecular biology.
Harvard University Press, Cambridge MA, pp 204-214
Morgan TH (1911) Random segregation versus coupling in
Mendelian inheritance. Science 34:384
Orgel LE, Crick FHC, Sapienza C (1980) Selfish DNA.
Nature 288:645-646
Penny D, Hoeppner MP, Poole AM, Jeffares DC (2009) An
overview of the intron-first theory. J Mol Evol 69:527-540
Pfeifer K, Tilghman SM (1994) Allele-specific gene
expression in mammals: the curious case of imprinted RNAs.
Genes Devel
8:1867-1874
Prabhu VV (1993) Symmetry observations in long
nucleotide sequences. Nucleic Acids Res
21: 2797-2800
Reanney DC (1978) Noncoding sequences in adaptive
genetics. In: Fox CF, Todaro GJ, Stevens JG (eds)
Persistent viruses.
Proceedings of the 1978 ICN-UCLA symposium on molecular and cellular biology
held in Keystone, Colorado, February 1978.
Academic Press, NewYork, pp. 311-330
Reanney DC (1979) RNA splicing and polynucleotide
evolution. Nature 277:598-600
Reanney DC (1984) RNA splicing as an error-screening
mechanism. J Theor Biol 110:315-321
Reese V (2002) Mutation repair: a proposed mechanism
that would enable complex genomes to better resist mutational entropy, and which
suggests a novel function for meiosis. The Human Behavior and Evolution Society
14th Annual Meeting, Rutgers University. Abstracts of presentations to session
on "New Developments in Biology," June 21, p. 40
Robertson M (1977) Immunoglobulin genes and the immune
response. Nature 269:648-650
Rogozin IB, Carmel L, Csuros M, Koonin EV (2012) Origin
and evolution of spliceosomal introns. Biology Direct 7, 11
doi:10.1186/1745-6150-7-11
Rudner R, Karkas JD, Chargaff E (1968) Separation of
B. subtilis DNA into complementary
strands. III. Direct analysis. Proc Natl Acad Sci USA
60:921-922
Tiemeier DC, Tilghman SM, Polsky FI, Seidman JG, Leder
A, Edgell MH, Leder P (1978) A comparison of two cloned mouse β-globin genes and
their surrounding and intervening sequences.
Cell 14:237-245
Tomizawa J (1984) Control of ColE1 plasmid replication:
the process of binding of RNA I to the primer transcript.
Cell
38:861-870
Vinogradov AE (2001) Within-intron correlation with
base composition of adjacent exons in different genomes.
Gene 276:143-151
Wilkinson DM, Ruxton GD (2012) Understanding selection
for long necks in different taxa. Biol. Rev. 87:616-630
Witkowski JA (1988) The discovery of 'split' genes: a
scientific revolution. Trends Biochem Sci
13:110-113
Yang S, Yuan Y, Wang L, Li J, Wang W, Liu H, Chen J-Q, Hurst LD, Tian D (2012) Great majority of recombination events in Arabidopsis are gene conversion events. Proc Natl Acad Sci USA 109: 20992-20997
Zhang C, Xu S, Wei J-F. Forsdyke DR (2008a)
Microsatellites that violate Chargaff's second parity rule have base
order-dependent asymmetries in the folding energies of complementary DNA strands
and may not drive speciation.
J Theor Biol
254:168-177
Zhang C, Li W-H, Krainer AR, Zhang MQ (2008b) RNA
landscape of evolution for optimal exons and intron discrimination.
Proc Natl
Acad Sci USA 105:5797-5802
Zuker M (1990) Prediction of optimal and suboptimal
secondary structure for RNA. Meth Enzymol 183:281-306
![]()
End Note (Feb 2013) Help from handing editor
So much of the early intron
literature having been in Nature,
this paper was first submitted there (17 May 2012), but was declined for
review (22 May 2012). It was then submitted to
Biological Theory,
where there were initially two conflicting anonymous reviews, one for
and one against. The handling editor then tried, with some difficulty,
to find new reviewers. On consulting those who might be able to advise
on possible reviewers, he later commented that: "In the process I
discovered a lot about the sociology of your field!
End Note (Nov 2014) Are introns a burden?
Sadly we learn of the unexpected death of the above handling editor (Werner Callebaut, born in 1952), in early November 2014.
In March 2014 BMC Evolutionary Biology published an interesting paper on the topic of "intron burden" (Gorlova et al. 2014). The view that human introns are a "burden" was held to be supported by a negative correlation between gene expression and total intron size or number within a gene, with high expression being deemed as something positive and low expression being deemed as something negative. But, even if these premises are accepted, the curves are bimodal. Figure 3a of the paper, for example, shows that as intron number increases from zero to three, the expression level of the corresponding gene increases dramatically. The correlation is distinctly positive. Following the authors' line of reasoning, this can be interpreted as showing that introns are beneficial, but as their length or number within a gene exceed certain limits, a possibly independent detrimental effect, much less evident at lower lengths or numbers, intervenes. In other words, when intron number or length exceed 3 or 5 kb, respectively, their presence in a gene is sustained despite the association of decreasing gene expression as the number or length grow. Thus, the benefits of introns could be very great. Genes that could not weather this presumed detrimental effect would have had to shed or shorten introns.
Gorlova et al. also note a positive correlation of intron size and number with evolutionary conservation of a gene (Fig. 2 of their paper). This correlation was most dramatic over the lower range of intron length and number. The positive correlation is consistent with observations in bacteria (usually without introns) and yeast (with few introns). Here, conservation (low evolution rate) correlates with high expression level (the so-called expression-evolution rate anticorrelation). Thus, the data from microorganism and human genomes are in agreement in the case of genes with few or short introns. Over this range, as intron number and length increases, conservation increases. The possibility that introns might have aided that conservation was not considered.
"Burden" is a loaded term, implying that net costs
might outweigh any benefits. Genes that are conserved must either depend
on efficient natural selection to eliminate organisms with mutations,
or be accurately corrected when they
mutate. For genes that are not conserved, the luxury of
accurate correction would seem less pressing. Thus, if introns were
concerned with maintaining genome integrity, then more intron "burden"
in conserved genes would be expected. On the other hand, in
extremis, there are the rarer positively selected genes, which vary
rapidly in amino acid sequence (Gorlova et al. assess conservation at
the protein sequence level; "CI values"). By virtue of these rapid amino
acid changes, such genes are favored by natural selection and
accumulate mutations that Gorlova et al. refer to as "functional
polymorphisms." For positively selected genes, these amino acid changing
mutations do not imply any lack of "functional significance" or
"functional importance." But here the error-correcting role can
rely less on exonic synonymous sites, so any error-correction that is
required comes to depend more on introns, which can then appear more
conserved than exons (see Fig. 4 in above paper). Then the intron
"burden" could increase.
Genes that are conserved over long evolutionary time periods
(presumed to reflect their functional importance) would seem to have had
more opportunity to resolve their
conflicts with other forms of information that need to pass
through the generations in the same genomes. Such forms include
stem-loop pressure, GC-pressure and, for exons, AG-pressure ("R
loading"). Sometimes synonymous substitutions [SYN] would suffice and
protein sequence conservation (low chain-terminating [NON] and missense
[MIS] base substitution values) would be apparent. Thus, the NON/SYN and
MIS/SYN base substitution ratios would be low, and intron number/length
could be high. But if high synonymous values and increases in intron
number/length were insufficient, amino acid changes (non-synonymous
mutations [MIS]) might be necessary for resolving conflicts. For
example, an exchange of arginine for lysine (both basic amino acids)
might suffice. Or more extreme exchanges might be required, ultimately
extending to chain-terminating [NON] mutations. In this circumstance,
both MIS and SYN values can change and intron number/length can be less.
Thus, there could be "a
significant negative correlation between MIS/SYN and the number of
introns (R = −0.05, n = 3,363, P = 0.006)."
![]()
Return to: Introns pages (Click Here)
Go to: Bioinformatics Index (Click Here)
Go to: Homepage (Click Here)
Go to: VideoLectures (Click Here)
![]()