|
Patterns in codon usage of different kinds of species
Richard Grantham (1980) Trends in Biochemical Sciences 5, 327-331. (With permission of the author, and copyright permission from Elsevier Science; Click Here)
Current evolutionary debates involve sociobiology, neutralism and origins of different kinds of genomes. Sociobiology and neutralism can be seen as opposing themes. The first proclaims that the phenotype and its comportment are the products of gene structure (1,2). But neutralism assigns a minor evolutionary role to molecular changes in the gene (3). As for genome origins, the monophyletic substructure of life has been upset in the last few years by observations on mycoplasmas, bacteria, mitochondria, plastids and viruses(4-6). I believe investigations into the way that the code is exploited in various species can throw light on all these questions. Consequently, my justification for all this sequencing is that nucleic acid sequences reveal how the code is working, or has been worked. There is of course interaction in each of the above debates between the research methods used and the results found. For example, neutralism has been based on studies of amino acid substitutions and the results have been extrapolated to molecular evolution as a whole. Kimura says that:
An independent view of evolution will be exposed here. My evolutionary outlook derives from work with a new kind of methodology, based on nucleic acid sequences, that my colleagues and I have developed in recent years. We state our main result as a hypothesis because further testing is required to establish its general validity: all genes in a genome, or more loosely genome type, tend to have the same coding strategy. By this we mean they employ the codon catalog similarly; that is, they show similar choices between synonymous codons, or between degenerate bases (those in codon position III). Hence a systematic exploitation of the code's degeneracy, particular to the genome type, is portrayed in each gene sequence. Unlike the picture emerging from studies on proteins with the same method (see below and Refs 7-9), our results with nucleic acids resemble classical systematics by distinguishing groups of like species. For example, the most gross observation is that viruses and mammals have widely separate coding strategies. This is evident by simple comparison of codon frequencies in the two kinds of genes.
To eliminate the influence of amino acid frequency on codon frequency, consider only the eight sets of codons called "quartets" (see Fig. 1). Each of these 32 codons belongs to a set of four synonymous triplets in which only the third base varies. Thus a complete choice of bases exists for filling codon position III without changing the resultant amino acid. This simplified approach gives only a partial view of the functioning of the code since there are 29 other amino acid codons, but we have found that the pattern is quite similar to that obtained with all 61 codons (7-9).
Fig. 2 shows the composition of the third bases of these quartets for 119 mRNAs taken together. We see that pyrimidines are generally preferred to purines as degenerate bases. Fig. 3 portrays systematic differences between genome types in filling codon position III. Thus quartet third bases in mammalian messengers contain less A and less U but more C and more G than in mRNAs of any other genome type. Little overlap in coding strategy occurs among individual genes of different genome types (7). The degenerate base choices in each mRNA consistently characterize the genome type of the relevant gene.
In the above comparison, mRNAs for mouse immunoglobulins (Ig) were excluded from the data for other mammalian mRNAs. Ig mRNAs use a sub-strategy in which an average of only 47.3% C+G is found in quartet position III while the other mammalian messengers show 70.9%. Also mouse Ig mRNAs use three times as much A as other mammalian messengers. The frequencies of C and U are close for general mammalian mRNAs and Ig mRNAs; the difference mainly lies in the use of purines. Thus in quartet position III, Ig mRNAs have a G/A ratio of only 0.6 while other mammalian messengers have a ratio of 4.0. The Ig coding strategy, unlike that of other mouse mRNAs, curiously resembles that of papova viruses (6-9). Of all the sequences so far obtained. mammalian messengers (excluding Ig messengers) repeatedly exhibit the highest C+G content and the lowest A content in degenerate bases (7). Another aspect visible in Fig. 3 is the variation in use of A versus U, and C versus G. Five times more U than A appears in quartet third bases of mRNAs of single-stranded DNA phages (this of course increases the contrast between U and A in Fig. 2. since 35 of the 119 total mRNA sequences come from ssDNA phages). Conversely, all groups show fairly even use of C and G except Ig, whose mRNAs have over twice as much degenerate C as G. Correspondence analysis provides gene distances A better image of the genome hypothesis is to be had by the simultaneous consideration of all 61 codons in the total sample of mRNA sequences. The best tool we have found for demonstrating this is correspondence analysis, which is a multivariate method adaptable to assessing biological variability and allowing graphical representation of the quantitative results (10,11). The analysis identifies and measures the importance of the various factors in codon usage that separate mRNAs. Variation of the frequency, among all mRNAs of each of the 61 codons is simultaneously calculated; the results position each messenger as a point in a multidimensional space. Then the data are projected on to a plane whose horizontal and vertical axes correspond to the first and second most important factors, respectively, in creating distance between the mRNAs. Grouping is achieved by the automatic classification method of Fages (12), which is equivalent to minimizing the variance in each class of a chosen number of classes. Some distortion in the projection is inevitable but this does not affect the classification. Two neighboring mRNAs in the plane can belong to different classes if the perpendicular distance between them is great. This means that factors other than the first two are important in distinguishing their coding strategies.
Results of correspondence analysis on 119 mRNAs appear in Fig. 4, in which separation of classes (delimited by automatic classification) is highly correlated with genome types. Two new groups, having too few total codons for inclusion in Fig. 3, are yeast mitochondrial, and yeast and slime mold genes. The seven Ig messengers lie between the upper right tip of the mammalian group and the top of the PAB group (papova, adeno and hepatitis B viruses). The double-stranded DNA bacteriophages occur mainly between bacteria and the large single-stranded DNA class. However, neither the Ig nor double-stranded DNA phage mRNAs constitute a separate class in this analysis. Messengers furthest to the left contain 88-90% C+ G in quartet position III while those furthest to the right have only 3-10%. There is little contamination of classes by genes of a different genome type (see Ref. 7 for identification and placement of each of the 119 mRNAs). This approach does not simply reproduce classical systematics; the figure contains new information on evolutionary mechanisms and paths. Nevertheless. it does sort genes according to genomic origin; therefore, it demonstrates that evolutionary change in genes is related to the differentiation of taxa. We wondered of course, how much the mRNA correspondence analysis pattern of Fig. 4 depended on the proteins coded. A correspondence analysis coupled with automatic classification was therefore done on the frequencies of the 20 amino acids in the 119 proteins; this analysis is shown in Fig. 5. No correlation between Figs 4 and 5 has been found. Indeed, we have not been able to account for placement of the proteins in Fig. 5. Viral, bacterial, mammalian and other proteins often lie in the same class. Every one of the seven classes of Fig. 5 includes proteins of viruses and at least one other genome type. We conclude that mRNA sequences contain other information than that necessary for coding proteins. This other "genome-type" information is mainly in the degenerate bases of the sequence. Consequently, it is largely independent of the amino acids coded (see Refs 7-9).
Explaining variations in coding strategy Why should individual genes segregate according to genome or genome type as in Fig. 4? One possible reason is metabolic discrimination between nucleotide bases. The basis for the mechanism would be an evolutionary interaction between concentrations of mononucleotide pools and replication errors. Thus different species, or kinds of species, would have arrived at different optimizations of the tolerated error level and amount of each base in the pool. Theoretical and experimental work supporting this approach has been done by Ninio and colleagues (for example see Ref. 13). An error with a given base relates not only to its concentration in the pool but also to that of the adjacent base. The error depends both on the time available for incorporation and for proof-reading. Incorporation time is a function of the concentration of the base being incorporated, while time for correction depends on concentration of the next base in the sequence. If the pool contains an abundance of the next base it will be incorporated rapidly, leaving little time for proof-reading of the first base. The mononucleotide pools have not been measured for all tissues and cells, hence correlation with the gene pattern in Fig. 4 has not been tried. A second possibility is regulation of replication or transcription through the choice of degenerate base. The speed and accuracy of copying could be influenced both by the nature of the base and its relative concentration in the pool, without invoking a proof-reading mechanism. Taxonomic groups could have exploited this double lever in varying manners, leading to different degenerate base compositions in the genes. Of course. this notion has implications for untranslated regions also, but lack of data precludes one from deciding on its applicability. The optimization of secondary structure by choices between possible third bases might also affect coding strategy. The optimal secondary structure for a messenger could depend on cell size, nuclease content, salt concentrations, temperature range and other factors. In addition, the form of the messenger could be a brake to control its translation rate. Unfortunately. progress has been slow in determining mRNA conformation in the cell experimentally. Another explanation for the genome type distances of Fig. 4 might be that the codon and anticodon populations are harmonized. Here we encounter a problem with regard to parasites. E. coli is a human symbiont and phages are E. coli parasites. If nucleotide pool concentrations are the determining factor in the separation of mRNAs revealed by correspondence analysis, parasites and hosts should have similar placements in the figure. The two examples are not analogous, however: E. coli cells establish their own pools. Coliphages do not, and hence they might be expected to have a coding strategy closer to E. coli than E. coli has to man. Curiously, bacteria fall about halfway between human and single-stranded DNA genes, although highly expressed bacterial mRNAs are nearer the large single-stranded DNA class. The double-stranded DNA phage messengers are closer to bacterial mRNAs (7). Why should single-stranded DNA phages (fxl74, G4, M13 and fd) fabricate messenger sequences that use the translation apparatus and tRNA of their bacterial hosts, yet make different choices from the host among synonymous codons? The host has had a long time to harmonize codon and anticodon populations. This may indicate that single-stranded DNA phages are relatively recent invaders of bacteria and have not yet evolved codon frequencies perfectly adapted to the bacterial anticodon distribution. Of course, a too-perfect adaptation could mean extinction through killing too many bacteria. However, the mRNAs of double-stranded DNA lambdoid phages are near those of bacteria; this could mean they have been bacterial parasites for a longer time. Another problem is mitochondria. Yeast mitochondria genes fall about as far from yeast genes as papova virus genes do from human genes (we shall soon begin work with human mt sequences). The coordinated use of codons and anticodons is discussed further in Ref. 6 where it is shown that the mammalian cell must be deficient in tRNA for translating the frequent A-ending codons of SV40 mRNAs. It is easy to imagine that this is a reflection of the relatively slow growth of papova viruses in primates, but the subject needs further analysis and experimentation. Indeed, the overall strategy of papova viruses is obscure. SV40 is found in all tissues of monkeys. Although these viruses are considered neurotropic. They can transform lymphocytes (the site of production of Ig mRNAs). As seen in Fig. 2 of Ref. 7, mRNAs of papova viruses have coding strategies closest to those of three Ig among all mRNAs sequenced in mammals. Hence it would be interesting to know the tRNA distribution in lymphocytes. Another curious aspect of papova viruses is their 'poly A tendency'. Of the above 119 messengers, 19 exhibit frequent runs of four or more adenines ( 4.0% of total bases). Of these 19, five are SV40 or BKV mRNAs (14). Thus their elevated content of degenerate A is at least partly a reflection of poly A tendency. These five papova genes use much more A and U in codon position III than do those of mammals (see Table 5 of Ref. 6), except for mRNAs of these Igs and three hormones, which also fall in the same class with papova viruses (7). None of the six Ig or hormone messenger sequences shows poly A tendency however. Poly A tendency determinations should help to understand differences in coding strategy in these and other genes. Nonetheless, we have not yet been able to 'rationalize' the vertical axis of Fig. 4. Finally, third base choice could regulate the expression of mRNA at the translation level (7-9). The mRNAs of abundant proteins lie at the bottom of Fig. 4, whose vertical axis is therefore linked to mRNA expressivity. Such a regulation might be realized by controlling the secondary structure of the messenger. However, the explanation appears less simple. Codons in the class of highly expressed bacterial genes have less C and G in position III than do those of other bacterial genes (note that as well as being lower in the figure, the highly expressed mRNAs are to the right of other bacterial mRNAs). But the axis representing degenerate C + G content, which should be closely related to variation in secondary structure, is horizontal not vertical. Hence we must consider other possibilities of mRNA regulation. It is conceivable that third base choice is constrained by the relative concentrations in the pool of the four monoribonucleotides and that there is an optimum choice of bases for maximizing the rate of mRNA transcription (or avoiding errors). Thus the number of copies transcribed of each messenger may be influenced by the third base composition relative to these concentrations. However, the existence of such a mechanism would not prevent another control at the translation level. A possibility for translation regulation exists in codon context effects. It has been experimentally demonstrated that the interaction of tRNA with mRNA is not independent of mRNA sequences outside the codon. Recent results suggest that any given codon may be read preferentially by one or another member of an isocoding tRNA family, depending on the context (neighboring codons). The efficiency of reading a particular codon can vary over a ten-fold range (15). Consequentlv, 'internal' regulation of translation of a messenger would be possible through degenerate base choices (7-9). Evolutionary interaction with the monoribonucleotide pool concentrations could exist to optimize the overall cell economy. As already shown, substitutions in protein are highly correlated with physicochemical properties of the exchanging residues (16). These exchanges, however, are not all there is to evolution or even molecular evolution. The nature of the protein coded has little to do with the position of its messenger in Fig. 4 (compare Figs 2 and 3 of Ref. 7). The different coding strategies can be viewed simply as distinct ways of coding a given protein. For example, the average protein of Dayhoff (17) could be coded by an mRNA falling in any one of the classes of Fig. 4. But if that protein, or any other, is to be produced by a species belonging to a genome type represented by one of these classes, I predict that its mRNA will make choices among synonymous codons such that the position of the messenger given by correspondence analysis will be inside the class of its genome type. As seen above, such predictions pertain to most genes in a genome or genome type, but a few exceptions do exist. These results also imply that we now have a means of estimating, before sequencing either the mRNA or the protein, the degenerate base composition for mRNAs of proteins of known origin and amino acid composition. Consequently, the total base composition of the messenger can be predicted since the non-degenerate bases are decoded without ambiguity.
The systematics of viruses, bacteria, mitochondria and of small species and genomes in general is difficult, partly because there is less phenotype to work with and systematists have often worked exclusively with phenotypes. Our ideas about the origins of theme genomes, and whether they are autogeneous or endosymbiotic, are being revised (4). The genome-distance-by-coding-strategy approach can aid in resolving such questions. As the sample of sequenced genes and genomes grows our analyses can be refined and the number of classes in the correspondence analysis increased. The genome hypothesis resulted from studying codon usage in the mRNA in our sequence bank. Additional analyses on the same sequences have been done or are in progress. We are finding further examples of differences and similarities between genome types, genomes and genes. This work continues to indicate protein-independent molecular evolution of a non-neutral character, and may aid in understanding and extending the genome hypothesis. References 1 Wilson, E. 0. (1978) On Human
Nature, Harvard University Press, Cambridge MA
Patterns in codon usage of different kinds of species RICHARD GRANTHAM, PASCALE PERRIN, AND DOMINIQUE MOUCHIROUD (1986) Oxford Surveys of Evolutionary Biology 3, 48-81 (With permission of the first author, and of Elizabeth Mann for Oxford University Press) 1. Introduction When Miescher discovered nucleic acids in hospital pus in 1869, a decade after publication of Darwin's Origin and just following Mendel's experiments, the development of molecular evolution became possible. Recognition that DNA was the genetic substance, however, had to wait another 75 years. Of course the biochemical and statistical methodologies were lacking, but around 1872 Galton began introducing statistical methods into biology. Such methods are necessary for arriving at reliable generalizations. Galton, and in the next decade Weismann and others, also did experiments that contributed greatly to the evolutionary synthesis a half-century later. Although Darwin was aided by personal contacts with Lyell, Huxley, and Galton, isolated and abbreviated careers were the lot of Mendel and Miescher, and their work was not followed up for many years after their deaths. Partly as a result of this, perhaps, molecular evolution as a discipline has not fully established itself. We do not yet have a theory of molecular evolution and remain largely at the stage of data gathering. Articulation between biochemical phenomena and genetic expression in populations is poorly understood and hypotheses, when they can be formulated, are often difficult to test.
Weismann would rejoice. For example, for coding leucine, which has six codons, mRNA of human mitochondria prefers CUA, that of nuclear genes of certain plant species CUC, of human nuclear genes CUG, of ssDNA bacteriophages G4 and j X174 CUU, of AIDS virus UUA, and of yeast nuclear genes UUG (Anderson et al. 1981; Ikemura 1985; Ratner et al. 1985; Roe et al. 1985; deBoer and Kastelein 1986; Li et al. 1985; GenBank release 35).
Analysis of overall codon usage by different taxonomic groups has remained a marginal activity for two reasons.
This review seeks to summarize and interpret the main features of variation in selection among synonymous codons. Why codon usage in each species is biased is not known. Nor are we sure that a general bias for the whole biosphere exists, because the sample of all sequenced genes is still too small. Some hypotheses have been announced, but it is often not clear whether one should expect the bias in each species to be determined by phenotypic or genotypic considerations. A tangle of proximate and ultimate causes, and of cause and effect ambiguities, is encountered. For example, what is the influence on a species' use of the codon catalogue of food, population size, niche, individual lifespan, or size of the phenotype? The response seems to be 'none' each time. In fact, we are simply not ready to answer these questions. In this paper we take the view that coding strategy is a fundamental evolutionary structure and that species or kinds of species can be characterized by variation in this structure. Indeed, certain distinctive patterns have been reported. Three recent reviews have aided in preparing this synthesis (deBoer and Kastelein 1986; Ikemura 1985; Li et al. 1985). We have selected 10 species groups for special study; these are the groups with the greatest number of published gene sequences. 2. Interspecific patterns of codon choices Only part of the [conventional protein-encoding] information contained in the genotype is expressed in the phenotype as protein. This part varies from over 90 per cent for small viruses to only about 2 per cent in humans and other mammals. Another quantitative genetic difference between species is in degenerate base use.
Analysis of all sequenced genes for overall use of the 61 codons separates them into groups of similar species. For example, a correspondence analysis on the first 54 mRNA sequences published for eukaryotes showed separation between yeast mitochondrial and yeast nuclear genes, and between fungus and animal genes (see Fig. 16 of Grantham et al. 1981). In another correspondence analysis, human and yeast nuclear genes and their mitochondria were seen to have distinct coding strategies (Grantham et al. 1983). That is, there are patterns of usage of the codon catalogue. These graphic patterns have been accompanied by identification and quantification of the importance of the principal factors responsible for the separations between messengers observed.
2. 1. HUMANS AND OTHER VERTEBRATES Although total human nuclear DNA, like that of other mammals, contains about 41 per cent (G + C), all major families of protein coding sequences have over 50 per cent (G + C) in degenerate bases (see Figs 5 and 7 of Grantham et al. 1985). In fact C-ending codons are favoured in 14 of the 16 possibilities for choice between such codons and those ending in other bases, while keeping the same amino acid each time. The two exceptions are CUG and GUG, the codons of highest frequency for leucine and valine, respectively (Grantham et al. 1981; Li et al. 1985). G-ending codons of Thr, Pro, Ala, and Ser are rare because they have C in position 11, forming the di-nucleotide CG, which is strongly avoided in man and most eukaryotes (see below). Why C-ending codons generally predominate in vertebrate sequences over those ending in A or U is not clear. To appreciate this, note that the complementary triplet for AAA is UUU and that for GGG is CCC; since G-C pair formation liberates much more free energy than A-U pairing, the pairing of the last two triplets is called 'strong' binding and that of the first two triplets 'weak' binding. Consequently, as seen below, UUC, AUC, UAC, and AAC are expected to be used more frequently than their U-ending cognates from codon-anticodon binding energy considerations. These four codons form pairs with their specific anticodons characterized by intermediate energies while their cognates UUU, AUU, UAU, and AAU form pairs of weak interaction energy. That is, in each of the four cases the anticodon is the same for both the C- and U-ending codon and it contains G in the degenerate (wobble) position; G forms a much stronger bond to C than to U. On the other hand, elevated frequencies would not be expected, given the overall genome composition, for triplets CCC, GCC, CGC, or GGC, which form extreme energy pairs with their anticodons (Grantham et al. 1981). But, the four latter codons, like the four former C-endings ones, are each of highest frequency within their specific set (Grantham et al. 1981; Ikemura 1985; Li et al. 1985). When a (methylated or otherwise) modified base occurs in the anticodon wobble position, as happens frequently in these eight cases (Sprinzl et al. 1985), we do not understand why C is favoured over U as third base. This field of research has been neglected for several years and no good explanation has been found for the tendency to high G + C content in codon position III of most genes. Pairing energies involving modified bases have not been quantified. Adams and Eason (1984), and Perrin (1984) have proposed that mutation rate decreases with increasing G + C content, which would tend to stabilize coding strategy. However, confirmation of this notion has not appeared in the case of CpG mutating to UpG (Cooper and Gerber-Huber 1985; Grantham 1985). 2.2. INVERTEBRATES Invertebrates will be exemplified by two species, the nematode Caenorhabditis elegans and the fruit fly Drosophila melanogaster. Codon choices differ strikingly between the two species. For example, in the nine highly expressed genes of C. elegans sequenced (Kramer et al. 1982; Files et al. 1983; Karn et al. 1983; Klass et al. 1984; Spieth et al. 1985), CUU is the Leu codon of highest frequency while CUG is favoured in the 46 Drosophila sequences. Furthermore, avoidance of doublets CG and UA is much more severe in the worm. As seen below, a rather strong case for energy optimization in codon-anticodon pairing can be made with C. elegans. 2.3. YEAST Since deBoer and Kastelein (1986) have just summarized codon frequencies in 34 yeast genes, we take their data for comparison with other species. As appears in Section 3, CpG avoidance in Saccharomyces cerevisiae and Homo sapiens is similar; however, UpA avoidance in yeast is stricter than in man, being surpassed only by that in C. elegans among species studied here. No good explanation for avoidance of the UA doublet has appeared. Codon-anticodon pairing energy optimization in yeast has been discussed by several authors, who have found a strong preference for middle-level energies in highly expressed genes (Bennetzen and Hall 1982; Ikemura 1985; deBoer and Kastelein 1986; Li et al. 1985). In summary, overall usage of the codon catalogue in genes for abundant proteins is such as to assure intermediate levels of codon-anticodon interaction energy, in yeast as well as in E. coli. 2.4. PLANTS AND CHLOROPLASTS Of the amino acids having codon choices, only Gln favours the same codon, CAA, in chloroplasts and plant nuclear genes sequenced, as can be seen below. This suggests different origins for these two plant genomes. Chloroplasts appear to have more genetic freedom at the molecular level than do mitochondria. Eleven of the 18 amino acids show highest frequency for the same codon in nuclear and mitochondria genes of man (insufficient plant mitochondria have been sequenced for a good comparison). It is also intriguing that 10 preferences are the same between plant nuclear and E. coli genes, making it difficult to believe that chloroplasts descended from Eubacteria since preferences coincide in only five cases between chloroplasts and E. coli. Of these 18 amino acids, 11 show the highest frequency for the same codon in man and E. coli (see below). We are therefore a long way from understanding what conserves and what changes codon preferences. As will be revealed in Section 3, chloroplast genes so far sequenced favour UUA for coding Leu. They weakly avoid CpG and even more weakly, UpA. They have much higher frequencies for A and U than C and G as degenerate bases and show no evidence of pairing energy optimization by C/U or A/G choices (Boudraa 1987). CUC is slightly favoured over UUG as preferred Leu codon in plants, as seen below. 2.5. MITOCHONDRIA The complete genomes of Xenopus laevis, mouse, rat, bovine, and human mitochondria have been sequenced; each contains 13 long, open reading frames, that is, potential coding sequences for proteins free of terminator triplets (Anderson et al. 1981, 1982- Bibb et al. 1981; Saccone et al. 1981; Roe et al. 1985). In some cases the protein has not been identified, hence these open reading frames in the genome sequence are potential genes, most of which, however, have been found to correspond to functional proteins.
For example, human mitochondria generally favour codons ending in C while Xenopus mitochondria have higher frequencies for those with U as third base. Hence, the amphibian mitochondrial system prefers G-U wobble to the standard G-C reading of codon position III found in mammalian mitochondria (Roe et al. 1985). Mitochondria thus present a curious evolutionary history. From Drosophila to man their genome size seems minimized and varies little. Gene order is different between Drosophila and vertebrates, but practically identical from X. laevis to man (Roe et al. 1985). Also, codon use differs greatly between mitochondria of X. laevis and man; 13 amino acids have different preferred codons between the two species. Between mitochondria of Drosophila and Xenopus, nine amino acids differ in codon preferences while between those of yeast and Xenopus 10 such differences exist. The only such difference between mitochondria of yeast (10 sequenced genes) and Aspergillus nidulans (12 sequenced genes) is with the amino acid Met, the former favouring AUG, the latter AUA (GenBank release 35). This suggests strong conservation of coding strategy in the two species over long times, although no date for their common ancestor has been proposed. Indeed, we do not know how human mitochondria evolve - that is, how they are and have been selected. Do they have to be evaluated at the level of the host phenotype? This seems unlikely in view of the values for certain indexes presented below, for to maintain these values would seem to mean the elimination of many host individuals at each generation.
3.1. SEQUENCE PHYSICOCHEMICAL CHARACTERISTICS The protein coded, of course, conditions properties of the nucleotide sequence, but much freedom for varying properties through degenerate base choice remains. Consider a few structural aspects and sequence properties:
All these structures probably interact with each of the properties; consideration of the evolutionary importance of these features has begun, notably with the work of Rich and colleagues (Johnston and Rich 1985; see also Temin 1985; Grantham et al. 1985). 3.2 TRANSLATION OPTIMIZATION Do codon frequencies adapt to tRNA concentrations or the converse (Garel 1982)? Both are adapting to something, that is, they are being selected. Changes among synonymous codons do not change protein structure, but they may influence the amount of protein made and the efficiency of its synthesis. That is, rate of translation and quality of the product can both be controlled by codon choice because some triplets translate more rapidly and accurately than their cognates (a protein containing translation errors may have a different half-life and biological activity from a more faithful copy).
What such differences mean in evolution is still obscure. Clearly, there is harmonization between codon and anticodon intracellular populations in yeast and E. coli (Ikemura 1981, 1982, 1985; Bennetzen and Hall 1982; Gouy and Gautier 1982; Grosjean and Fiers 1982) and there can be little doubt that this facilitates translation. Codons of high frequency in mRNA are in general decoded by anticodons of high frequency in the cell's tRNA. This harmonization of the two intracellular populations optimizes translation by increasing its speed (since a high frequency codon is decoded faster, due to more specific anticodons being present in the cytoplasm) and decreasing mismatching errors (Gouy and Grantham 1980). The extent of selection on codon-anticodon pairing energies has not been generally studied; analyses have been confined to E. coli and yeast (Bennetzen and Hall 1982; Grosjean and Fiers 1982; Ikemura and Ozeki 1983; Ikemura 1985). We attempt an extension of this phenomenon to Metazoa, as shown below. 3.3. ANCESTOR SEQUENCE BIAS The life system started with certain sequences, possibly with one or a few particular sequences. It is sometimes thought that, because of the mutation process, all trace of the original sequences has been lost. But, as seen in this review, coding strategy appears to be conserved over long evolutionary time. In addition, even though the mutation rate is sufficient to wipe out the original condition, natural selection has probably been interacting all the time and perhaps re-selecting certain features of the starting sequence, although the function and environment of the sequence have changed. Many things have changed in the biosphere in the last 3.5 thousand million years, but many have remained rather constant (temperature, pressure, inorganic composition of the earth . . . ). It is reasonable to suppose that these relatively constant factors may be reflected in the conservation of certain sequence characteristics. We also believe that each lineage has developed its own strategy for codon choices and has had to contend with whatever bias existed in the ancestor sequences. In some cases the lineage may have adjusted to and conserved the ancestral bias instead of letting it mutate away (this is probably one function of repair enzymes). The above is only logical, of course, and we want to test this logic when possible. 3.4. CODON CHOICE INDEXES Several indexes requiring only codon frequencies and simple arithmetic aid us in assessing the importance of the three above influences, especially the second one. Absolute frequencies of the 61 codons in the 10 kinds of species appear in Table 1. Tables 2 and 3 then show values for the indexes in each kind of species and some mitochondria. The first two indexes, NCG/NCC and NUA/NUU, concern CG and UA doublet avoidance, and are explained in legend Table 2. The third kind of index relates to energy optimization in codon-anticodon pairing during translation; the explanation follows.
Degenerate C/U choice is interesting because ordinarily the same anticodon responds to synonymous codons ending in C or U. Interchanging A and G in codon position III, however, often implies changing the anticodon. C/U choices clearly relate to energy optimization in codon-anticodon pairing. Thus, the pattern of the choices should be indicative of the importance this parameter has had in evolution.
If energy optimization exists, codons having G or C in the first two positions should prefer A or U in position III. Likewise, codons with A or U in position I and II should tend to increase codon-anticodon interaction energy by choosing G or C as third base (Grosjean et al. 1978; Grantham et al. 1981; Gouy and Gautier 1982; Grosjean and Fiers 1982). Schematically, WWX codons, where W (weak binding) is A or U and X is any base, would prefer S (strong binding, that is, G or C) degenerate bases. Similarly, SSX codons would tend to use A and U as third bases. Middle energies provided by mixed doublets (MM: one W and one S base) serve as controls: MMX codons should show no systematic bias under this hypothesis. We must recognize at the outset that the eukaryote system has many more anticodons than do prokaryotes and that modified bases in the anticodon, which may sometimes change considerably the pairing energy, occur frequently. These changes have not been quantified, however, and consequently our analyses have been done without taking them into account. We compare C/U degenerate choice in codons of weak binding energy in the first two positions to those of strong binding energy in these positions. Frequency ratios of C- and U-ending codons are, respectively, represented by WWC/WWU and SSC/SSU. As explained above, these ratios are contrasted to that for codons having one W and one S base in positions I and II, MMC/MMU. Table 3 summarizes results on a few species and gene families.
The value in the last column of Table 3 reflects overall G + C content of degenerate bases, values in the first two columns are meaningful by comparison with that for MMC/MMU. We observe that the whole E. coli sample of 149 sequenced genes indicates translation pairing energy optimization but that the highly expressed (HE) sample of Gouy and Gautier (1982) shows much wider variation between the values for the first two columns. Because of anticodon base modification we cannot be sure that there is not general pairing energy optimization in man, but the data in Table 3 definitely imply its existence in the nine highly expressed genes of C. elegans and probably in Drosophila (where HE sequences are not separated).
We conclude that, in the absence of other explanation for Table 3, there is some codon-anticodon pairing energy optimization in Metazoa, at least in certain gene families, all the way up to and possibly including humans. These results, which are new for Metazoa, indicate that this phenomenon is linked to expressivity level, as in lower organisms (Gouy and Gautier 1982; Grosjean and Fiers 1982; Ikemura 1985). 4. Intraspecific variation and expressivity in the immune system 4.1. DESCRIPTION OF THE IMMUNE SYSTEM (For more on the immune system Click Here). The immune system of vertebrates is a complex organization involving several cell types and many protein molecules. Many of these molecules show a considerable degree of polymorphism which may be of two distinct types. (i) A classical multiple-allele polymorphism where the population as a whole shows a very wide range of phenotypes, but each individual expresses a defined, simple type inherited in normal Mendelian fashion by offspring. This is the case for the antigens of the major histocompatibility complex (MHC).
The MHC antigens provide a cellular context for foreign antigen recognition. A foreign antigen, e.g. a virus, presented on a cell is only capable of inducing an immune response under normal conditions if the responding cell shares MHC antigens with the presenting cell. The MHC antigens are the most polymorphic genetic marker known. (ii) A second and unique type of polymorphism is seen in the effector molecules of the B lymphocyte - the immunoglobulins (Ig) - and in the T cell receptors (Tcr). Every individual of a species expresses a vast number of chemically distinct molecules of Ig and Tcr. The molecular events which generate this variability are now moderately well understood. During lymphocyte differentiation, a rearrangement of the cell genome apposes a segment coding for the N-terminal portion of the final protein, via one or two junctional segments, to a position upstream of the region coding for an invariant C-terminal portion. The N-terminal (variable) region genes and the junctional segments are present in multiple copies, and the joining process has some positional flexibility; this leads to a combinatorial generation of many variant sequences. In addition, somatic mutations appear to increase the diversity of these segments during the life of the cell. Stimulation of a particular lymphocyte by its specific antigen leads to proliferation, producing daughter cells with the same genetic rearrangement, and hence to increased production of the relevant immune response. Both immunoglobulins and T-cell receptors are made up of two different polypeptide chains, coded at separate genetic loci, which both consist of variable (V) and constant (C) regions, leading to additional combinatorial variability. For immunoglobulins, the two polypeptides are called light chains (L) and heavy chains (H). Two different classes of L chains (Kappa and Lambda) are coded on separate chromosomes and possess distinct libraries of V regions. Either class may interact with any H chain to produce an immunoglobulin molecule. The several classes of H chain are coded at a single complex locus on another chromosome and share a common V region library. The different C region genes are arranged as a closely grouped series and each consists of several exons. The C region gene proximal to the rearranged active V region gene corresponds to IgM. The immature B lymphocyte expresses IgM from a mRNA generated by splicing from the V region segment (V-DJ) and the C region exons. Occasionally, a few molecules of other immunoglobulin classes may be made by the immature B lymphocyte by an alternative splicing event which removes the whole of the C coding segment together with the first intron. At a later stage in cellular differentiation, a further genome rearrangement may occur, leading to the elimination of the DNA coding for an arbitrary number of Ig C region genes and thereby bringing the V region coding segment with the first intron into apposition with a downstream C region segment. The B lymphocyte (and its progeny) will then produce a new class of immunoglobulin, but will conserve the L chain and H chain V regions, and thus the antibody specificity of the resulting molecule. This is called 'class switching'. These rearrangements (V-J joining, class switching) employ recognizable signal sequences in the genomic DNA as positional markers. In the following section we wish to examine whether the coding strategies within the different regions of these molecules may be involved in:
Similar studies on the less polymorphic molecules of the complement system, the Ig receptors with their nucleic acid and protein homologies to the immunoglobulins, and to the interleukins, etc., have been deferred for the present, due to the paucity of published sequence data. 4.2. DIFFERENTIAL MUTATION ALONG THE SEQUENCES In Ig sequences we observe differences in coding strategy between V regions and C regions (Perrin 1984). The most striking in terms of the 'genome hypothesis' (Grantham et al. 1980), is the variation according to segment type of percentage (G + C) in the third position of quartet codons (see legend Table 4). C regions use more C- or G-ending codons than V regions (Miyata et al. 1979; Perrin 1984). This appears to be a general tendency in vertebrates since A- and U-ending codons are rare in C regions of rabbit, rat, chicken, and caiman Ig genes (Perrin 1984). It is difficult to understand this phenomenon in terms of expressivity because C and V regions are transcribed on the same messenger.
The different specificities of antibodies are generated, in part, by recombinations between V and J (joining) segments of L chains, or V, D (diversity), and J segments for V regions of H chains, present in the germinal library (Tonegawa 1983). Somatic mutations, involving only V regions, help to increase the range of specificities (Bothwell et al. 1981; Gershenfeld et al. 1981; Perlmutter et al. 1984; Jerne 1985; Sablitzky et al. 1985). X-ray diffraction studies have shown that three zones of V regions are directly involved in antigen recognition. These are HV (hypervariable) zones. The rest of the V region constitutes the framework (FR). Gojobori and Nei (1984) revealed that HV domains have a nucleotide substitution rate three times greater than that in the FR. Are A- and U-ending codons used more in HV zones (Perrin 1984)? This appears to be the case.
The local (A + T) content seems to correlate with local nucleotide substitution rate. The lower (G + C) content of HV domains may lead to a less tight binding between DNA strands and thus increase the basic mutation rate (Adams and Eason 1984; Perrin 1984). It is known that replication accuracy changes along the genome (Bernardi and Ninio 1978). Preliminary analysis on Tcr coding sequences of mouse also indicates differentia1 usage of synonymous codons for V and C regions. But, the difference is smaller than in Ig segments and depends on the peptide chain. For example, for six b-chain sequences of murine Tcr (Chien et al. 1984; Hedrick et al. 1984; Patten et al. 1984; Saito et al. 1984) the values of (G + C)Q3 are 61.6 per cent for C regions and 46.3 per cent for V regions. We do not find differential codon usage between different domains of MHC sequences, which exhibit multi-allelic polymorphism, and not somatic mutation and segment recombination (Benacerraf 1981; Steinmetz 1984). 4.3. NUMBER OF DIFFERENT CODONS USED IN Ig GENES Harmonization between codon usage and tRNA availability occurs probably at the messenger level, as seen above (selection of tRNA genes may also take place, of course). The range of codons used in V and C regions is quite similar although relative frequencies of the different codons vary considerably between the two kinds of regions (Perrin 1984; unpublished observations). Analysis on codon choices in C g genes and C e genes has revealed no great variation (Grantham and Perrin 1985) in spite of their different contents in plasma. IgG represents 75 per cent of plasmatic Ig whereas IgE content is less than 0.1 per cent (Nisonoff et al. 1975), yet their codon usage appears similar. But, IgE may be highly produced locally, hence we cannot be sure its gene has not been selectively optimized for coding strategy. Therefore, so far no differential range in number of codons used has been found among the various Ig genes. The few qualitative data available on the tRNA lymphocyte population (Marini and Mushinski 1979) are too imprecise for a related study. 4.4. DISTRIBUTION OF CG, UA, UG, AND CA DOUBLETS, AND VARYING G + C CONTENT The 16 dinucleotides (doublets) differ in frequency in natural nucleic acids; this variation may be linked to regulation involving base modification (methylation). It happens that, in most eukaryote sequences studied, C followed by G is much rarer than C followed by any other base (Grantham et al. 1985). Vertebrate genomes are strongly methylated and C is the only base so modified. Cytosine is methylated only in CpG (Felsenfeld and McGhee 1982). The mC tends to mutate to thymine, raising (in RNA) the frequency of UG (and CA on the complementary strand in DNA) (Barker et al. 1984). CpG frequency is interesting for three reasons.
4.4.1. CG, UA, UG, and CA doublet frequencies in Ig coding sequences For this study we used a statistical test to compare observed and expected frequencies. The expected frequency is calculated by base permutation (Grantham et al. 1985; Gautier et al. 1985). Results are given in Table 5. They lead to three conclusions.
(i) CG doublets are avoided in human and mouse V and C regions. This avoidance appears also in V and C introns (unpublished results). (ii) The C regions (human and mouse) tend to avoid UpA, as V regions do, especially in position III-I (between codons). (iii) The C regions have more CA (except in III-I) and UpG (in all positions) than expected. The V regions also show this tendency, especially in position III-I. Since UpA frequency is lower than expected (either in all positions or in III-I), its avoidance cannot be explained exclusively by terminators being UA-beginning codons, as has often been suggested. In Ig coding V sequences, the avoidance of CpG increases from position I-II to III-I. C regions affected by this phenomenon contain high (G + C) content (>60 per cent in human C regions). Murine Tcr sequences also avoid CG and UA doublets and have elevated UG and CA frequencies in positions II-III and III-I (Table 6).
4.4.2. CpG frequency along the MHC sequences Studies on genes like HPRT (hypoxanthine phosphoribosyl-dehydrogenase) and G6PD (glucose-6-phosphate dehydrogenase) reveal CpG clusters in their 5' extremity (Wolf and Migeon 1985). CpG frequency varies along the MHC genes too (Tykocinski and Max 1984). Exons of each MHC sequence have been separated for two classes of histocompatibility antigen (MHC-I and MHC-II). Each exon codes for a determined structural domain of the protein chain (three domains in heavy MHC-I chains, two in a and b MHC-II chains). Table 7 gives results on CpG, UpA, UpG, and CpA usage, revealing the following.
(i) Exons (E2 and E3) for the first two domains of heavy MHC-1 chains show no avoidance of CpG, but do avoid UpA in position III-I; exon (E2) of MHC-II b-chains avoids UpA only in position II-III and does not avoid CpG in any position. (ii) Avoidance of both CpG and UpA in all positions occurs in MHC-I exon 4 and MHC-II a-exon 3 and b-exon 3. (iii) Exon 2 for MHC-11 a-chains avoids UpA and CpG in all positions except I-II for the latter doublet. CG doublet avoidance is similar in positions II-III and III-I of MHC genes. Translation constraints explain the variation in I-II. For example, exons coding for b-1 domains use slightly more quartet codons (70 per cent) than expected (4/6 = 67 per cent) to code arginine. Some exons that do not avoid CpG are rich in (G + C), but exons for the third domain of HLA-A3 and HLA-CW3 transplantation antigens (Sodoyer et al. 1984; Strachan et al. lQ84) have high (G + C) content (>60 per cent) while avoiding CG doublets. HLA-I 5' untranslated regions and the first two introns have expected CpG frequencies (Table 8), as does the HLA-AW24 5' extremity (N'Guyen et al. 1985).
4.4.3. Discussion The 5' regions of HLA-1 heavy chains, from the 5' end of the untranslated zone to the 3' end of exon 3 (5'UT + El + I1 + E2 + I2 + E3) do not avoid CpG. This may relate to the housekeeping status of classic transplantation antigens (Robertson 1985). These clusters in conjunction with hypomethylation may maintain gene activity (Wolf and Migeon 1985). But, this is not specific to HLA -I genes since we find CpG clusters in the 5' region of the b-chain sequence, too (HLA-II and H2-II). Adams and Eason (1984) proposed that stability of regions with high (G + C) content protects CG doublets against mutations via deamination, thus explaining non-avoidance of CpG. However, we have shown that some exons with high (G + C) content do avoid CG dinucleotides, for example, human Ig C regions. C regions seem to be highly methylated in non-mature B lymphocytes (Storb and Arp 1983). Other gene families show similar behaviour (Grantham 1985). Non-avoidance of CG (and UA) doublets occurs in the most polymorphic domains (Choi et al. 1983; Sodoyer et al. 1984). Exons for MHC-II a-1 domains (moderately polymorphic) avoid CpG less strongly than those coding a-2 domains (less polymorphic) (Benoist et al. 1983). Hence, a correlation between the degree of polymorphism and CpG frequency can be demonstrated. CpG clusters may assume a specific function. We know that, according to physiological conditions, nucleic acids may change in local conformation and that these changes are sequence dependent. A region rich in (G + C) under different conditions may assume B- or Z-DNA conformation (Hamada et al. 1982; Johnston and Rich 1985; Nordheim and Rich 1983). Z-DNA conformation may be a hot spot for rearrangement and gene conversion (Hamada et al. 1982; Nordheim and Rich 1983; Rogers 1983; Perrin and Grantham 1986). This scenario is compatible with conserving polymorphism. Gene conversion is a major mechanism for the generation of polymorphism in MHC genes (Weiss et al. 1983). Synonymous codon choices allow organisms or cells to vary doublet frequencies along the gene sequences. In turn the varying doublet frequencies could be linked to conformation changes between B- and Z-DNA, which could induce genetic variability and differential expression. Data are, however, still inadequate for definitely resolving the question of the relation between CpG frequency and expressivity. 5. Particularities in human viruses Human viruses in general have less G and C in codon position III than does the host genome, 47.5 versus 66.1 per cent, respectively, having been found in large samples (Grgntham et al. 1985). The viral genes also showed a larger variation that the host gene families in G + C degenerate content (see Fig. 5 of Grantham et al. 1985). In addition, the study revealed that DNA viruses vary more in coding strategy than do RNA viruses.
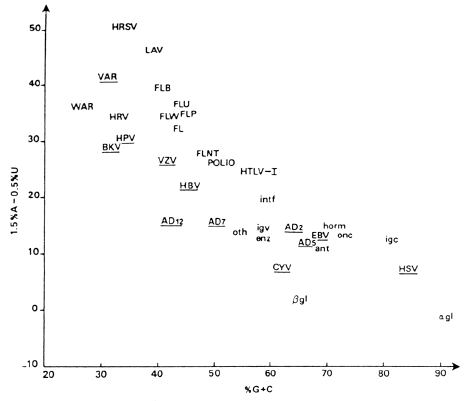
We now analyse 186 human and 243 virus gene sequences, each of at least 300 nucleotides. Table 9 groups the viral genes according to family, while Table 10 and Fig. 1 give percentage (G + C) of third bases in the sequences.
This larger sample confirms our previous findings: the 10 types of host genes in Fig. 1 all have a mean of over 50 per cent (G + C) in degenerate bases or in total composition (excepting interferons).
In all cases of host genes the degenerate percentage (G + C) is greater than that of total composition. Most viral genes have less than 50 per cent (G + C) in codon position III, although herpes EBV, HSV, and cytomegalovirus exceed this value, as do Ad 2 and Ad 5. Again we see that RNA viruses vary less in synonymous codon choices than do DNA viruses. The fast evolving influenza viruses reveal a surprisingly uniform percentage (G + C) in third bases. Overall, the new data confirm the previous conclusion that viruses do not closely imitate the use of the codon catalogue by the host. This is clearly portrayed in Fig. 2 (see Fig. 7 of Grantham et al. 1985), where the high variation of viral coding strategy compared to that of the human genome is also evident.
Contrasting AIDS virus (Ratner et al. 1985) to other retroviruses can be extended to codon choices. Five other retroviruses (BLV, bovine leukaemia virus; MoMuLV, Moloney murine leukaemia virus; AKV, strain AKR ecotropic endogenous murine leukaemia virus; RSV, Rous sarcoma virus; HTLV-1, human T cell leukaemia virus type 1) have been compared to AIDS. In summary (data not shown), for the three amino acids with six codons each and the five with four codons each, the preferred codon is nearly always different in all three viral genes (gag-pol-env) between AIDS and any of these five oncoviruses (Shinnick et al. 1981; Schwartz et al. 1983; Seiki et al. 1983; Herr 1984; Sagata et al. 1985). AIDS generally favours A-ending codons while these five viruses favour C- or G and, less often, U-ending triplets. Codons of highest frequency in AIDS for the eight amino acids are: Arg AGA, Leu UUA, Ser AGU, Thr ACA, Pro CCA, Ala GCA, Gly GGA, and Val GUA. These choices are consistently repeated in all three AIDS genes with only two exceptions. In env, UUG is slightly favoured over UUA for coding Leu and in gag, AGC and UCA are tied as highest frequency Ser codons. With any of the above five viruses, at most two of the eight amino acids show the same preferred codon as in AIDS for all three genes, and this occurs only with Arg and Gly in AKV, and MoMuLV. Much closer agreement in coding strategy is seen between AIDS and Visna lentivirus (VLV) (Sonigo et al. 1985). The preferred codon is identical for five of the eight amino acids in gag (VLV favours AGU for Ser, CCC for Pro and GUG for Val). With both pol and env genes all eight choices coincide between VLV and AIDS. Thus, the five other viruses appear evolutionarily distant from AIDS, as judged by favoured triplet for amino acids having full degeneracy in their codon sets. AIDS and VLV by this criterion are rather similar; this conclusion is compatible with other findings in suggesting that AIDS/LAV is more closely related to lentiviruses than to oncoviruses (Chiu et al. 1985; Sonigo et al. 1985). Table 11 summarizes codon use for the eight amino acids in the six viruses compared to AIDS. On the basis of absolute frequencies of preferred codons for these amino acids in the combined gag-pol-env genes of each genome, HTLV-l appears as most distant of any of the viruses from AIDS.
Shepherd (1982) has proposed that the present code derives from a prototype code in which purines predominated in codon position I and pyrimidines in position III, hence his 'RNY code' (R purine, N any base, Y pyrimidine). Indeed, for some reason the biological system prefers pyrimidines as degenerate bases (Grantham et al. 1983). Thus, with man, C + U in position III of the 195 genes of Table 1 is 55.4 per cent (52.3 is expected from the code structure). In fact, C is preferred over U as third base in human mRNA, as implied by the three columns of Table 3. This fact, unaccounted for by RNY theory (Shepherd 1982), apparently extends to most eukaryote organisms (excepting fungi), but not viruses (Grantham et al. 1983). It is not merely a consequence of CG doublet avoidance (avoidance of G as third base could tend to favour C) since Table 2 shows that CpG is favoured in codon position II-III of E. coli genes. From Table 1 we calculate that C represents 29.3 per cent of E. coli third bases while U only accounts for 25.5 per cent (human values are 33.5 per cent and 21.8 per cent). Since G is favoured (28.2 per cent) and A is avoided (17.0 per cent) as third base (human values are rather similar), a better primitive code model would be NNS (N, any base and S = G or C) for both humans and E. coli. In sum, the large gene samples we work with do not support the RNY hypothesis because it does not account for the asymmetry between C and U (or G and A) frequencies as degenerate bases. In addition, the apparent RNY working of the code in some species may relate to UpA and CpG rarity in codon position I-II. Both doublets are strongly avoided in yeast genes [see Table 2 above and entry 'Fun' (fungus) in Tables 13 and 14 of Grantham et al. 1985], on which Shepherd's model (1982 and 1984) was based. Their avoidance in position I-II, combined with the above general preference for pyrimidine third bases predicts the RNY (or RYY) schema. This is because CG and UA are both YR type doublets and the above avoidance necessarily favours A and G in position I. Note that UG and CA frequencies increase due to methylation of C in CG and mutation of mCG to UG (Bird 1980) and can compensate for CG avoidance, but not for UA avoidance. No molecular mechanism for explaining UA rarity has been advanced and no other YR type doublet has been proposed to be favoured by UA elimination. UpA is avoided in practically all kinds of sequences, both translated and untranslated, except mitochondria (Grantham et al. 1985). What could be done to further the understanding of bias in use of synonymous codons? We offer some speculative suggestions. One set of urgently needed data is concentrations of the different tRNAs that carry the same amino acid, the 'iso-acceptor-tRNAs'. Such data have been published only for bacteria and yeast (Bennetzen and Hall 1982; Ikemura 1985; de Boer and Kastelein 1986; Li et al. 1985), but their determination in various tissues of higher organisms and especially of man, for whom we now have many gene sequences for several protein families, would be most useful. This would allow assessment of the degree of harmonization between codon and anticodon distributions in different cells, both for nuclear genes and those of virus parasites. Thus, a better view of the evolutionary importance of this aspect of coding strategy would become possible. This appears especially cogent in understanding lymphotropic viruses, notably the AIDS virus (Grantham and Perrin 1986).
It will only become accessible as more data are accumulated on overall nucleotide metabolism, that is, the half-life and concentration in the cell of each kind of nucleotide, and perhaps that will only be a step in the right direction. It is already known that these factors vary widely in different cells, but no overall picture has been forthcoming. Perhaps a cell's overall nucleotide metabolism correlates with its degenerate base preferences, we can only speculate on this for the time being. We can, however, recognize a few related questions whose consideration may help in the general comprehension of the existence of this bias. (i) Why don't degenerate bases have the same composition as introns or other untranslated sequences? The provisional answer here is:
(ii) Why does each kind of transcription product (mRNA, rRNA and tRNA) have a rather limited range of G + C content that is most often different (and in animals, at least, generally higher) than that of the whole genome? The simplistic answer is that this is the way the biological system happened to develop, but there are probably other, functional and historical, reasons to be found. (iii) Why, for example, do a- and b-globin mRNAs make such different third base choices when they are translated at the same time and at similar abundances in the same cell? (iv) The same question can be asked regarding C and V segments of immunoglobulin mRNA. Here the situation is even worse since the two kinds of segments are incorporated into the same messenger. (v) Why is degenerate G + C content so high on the average and yet so variable in animal genes? Especially difficult to understand is the large variation in individual human genes, in which percentage (G + C) in codon position III runs from around 40 to over 90 per cent. These intraspecific codon biases must be maintained at great selective cost, most likely at the prenatal stage in our species, to eliminate mutants. Otherwise repair enzymes, for some unknown reason, would have to assure degenerate base use in each gene. As mentioned above, the selection of human mitochondria constitutes a similar problem. It is too easy just to say most mutations are neutral. The genome hypothesis has posed a chicken and egg dilemma whose resolution remains distant. Acknowledgements We thank M. Gouy, T. Greenland, J. L. Prato and D. Quilichini for unpublished data and help during preparation of the manuscript. References Adams, R. L. P. and Eason, R. (1984). Increased G + C content of DNA stabilises methyl CpG dinucleotides. Nucleic Acids Res. 12, 5869-77. Anderson, S., Bankier, A. T., Baffell, B. G., DeBruijn, M. H. L., Coulson, A. R., Drouin, J., Eperon, I. C., Nierlich, D. P., Roe, B. A., Sanger, F., Schreier, P. H., Smith, A. J. H., Staden, R., and Young, I. G. (1981). Sequence and organization of the human mitochondrial genome. Nature, 290, 457-65. DeBruijn, M. H. L., Coulson, A. R., Eperon, I. C., Sanger, F., and Young, I. G. (1982). Complete sequence of bovine mitochondrial DNA: conserved features of the mammalian mitochondrial genome. J. Mol. Biol. 156, 683-717. Barker, D., Schaffer, M., and White, R. (1984). Restriction sites containing CpG show a higher frequency of polymorphism in human DNA. Cell, 36, 131-8. Benacerraf, B. (1981). Role of MHC products in immune regulation. Science, 212, 1229-38. Bennetzen, J. L. and Hall, B. D. (1982). Codon selection in yeast. J. Biol. Chem. 2579 3026-31. Benoist, C. O., Mathis, D. J., Kanter, M. R., Williams, V. E., II, and McDevitt, H. 0. (1983). Regions of alielic hypervariability in the murine Aa immune response gene. Cell, 34, 169-77. Bernardi, F. and Ninio, J. (1978). The accuracy of DNA replication. Biochimie, 60, 1083-95. Bibb, M. J., Van Etten, R. A., Wright, C. T., Walberg, M. W., and Clayton, D. A. (1981). Sequence and gene organization of mouse mitochondrial DNA. Cell, 26,167-180. Bird, A. P. (1980). DNA methylation and the frequency of CpG in animal DNA. Nucleic Acid Res. 8, 1499-504. Blake, R. D. and Hinds, P. W. (1984). Analysis of codon bias in E. coli sequences. J. Biomol. Struct. Dyn. 2, 593-606. Boer, H. A., de and Kastelein, R. A. (1986). Biased codon usage: an exploration of its role in optimization of translation. In From Gene to Protein: Steps Dictating the Maximal Level of Gene Expression (eds J. Davis, B. Reznikoff, and L. Gold). Butterworths, New York. (In press.) Bothwell, A. L. M., Paskind, M., Reth, M., Imanishi-Kari, T., Rajewsky, T., and Baltimore, T. (1981). Heavy chain variable region contribution to the NPb family of antibodies: somatic mutations evident in a g 2a variable region. Cell, 24, 625-637. Boudraa, M. (1987). Variation de la strategic de codage dans le systeme vegetal. Genet. Sel. Evol. (in press). Chien, Y. H., Gascoigne, N. R. J., Kavaler, J., Lee, N. E., and Davis, M. M. (1984). Somatic recombination in a murine T-cell receptor gene. Nature, 309, 322-6. Chiu, 1. M., Yaniv, A., Dahlberg, J. E., Gazit, A., Skuntz, S. F., Tronick, S. R., and Aaronson, S. A. (1985). Nucleotide sequence evidence for relationship of AIDS retrovirus to lentiviruses. Nature, 317, 366-8. Choi, E., McIntyre, K., Germain, R. N., and Seidman, J. G. (1983). Murine I-A chain polymorphism: nucleotide sequences of three allelic I-A genes. Science, 22, 283-286. Cooper, D. N. and Gerber-Huber, S. (1985). DNA methylation and CpG suppression. Cell Different. 17, 199-205. Felsenfeld, G. and McGhee, J. (1982). Methylation and gene control. Nature, 296, 602-603. Files, J. G., Carr, S. and Hirsh, D. (1983). Actin gene family of Caenorhabditis elegans. J. Mol. Biol. 164, 355-375. Garel, J. P. (1982). The silkworm, a model for molecular and cellular biologists. Trends Biochem. Sci. 7, 105-8. Gautier, C., Gouy, M., and Louail, S. (1985). Non-parametric statistics for nucleic acid sequence study. Biochimie, 67, 449-53. Gershenfeld, H. K., Tsukamoto, A., Weissman, I. L., and Joho, R. (1981). Somatic diversification is required to generate the V genes of MOPC511 and MOPC167 myeloma proteins. Proc. Nat. Acad. Sci. USA, 78, 7674-7678. Gojobori, T. and Nei, M. (1984). Concerted evolution of the immunoglobulin VH gene family. Mol. Biol. Evol. 1, 195-212. Gouy, M. and Gautier, C. (1982). Codon usage in Bacteria: correlation with gene expressivity. Nucleic Acids Res. 10, 7055-7074. Gouy, M. and Grantham, R. (1980). Polypeptide elongation and tRNA cycling in Escherichia coli: a dynamic approach. FEBS Lett. 115, 151-155. Grantham, R. (1974a). Composition drift in the cytochrome c cistron. Nature, 248, 791-793. Grantham, R. (1974b). Amino acid difference formula to help explain protein evolution. Science, 185, 862-864. Grantham, R. (1980). Workings of the genetic code. Trends Biochem. Sci. 5, 327-31. Grantham, R. (1985). CG doublet difficulties in Vertebrate DNA. Nature, 313, 437. Grantham, R. and Gautier, C. (1980). Genetic distances from mRNA sequences. Naturwissenschaften, 67, 93-4. Grantham, R., Gautier, C. and Gouy, M. (1980a). Codon frequencies in 119 individual genes confirm consistent choices of degenerate bases according to genome type. Nucleic Acids Res. 8, 1893-1912. Grantham, R., Gautier, C. and Gouy, M. (1983). The genome as unit of selection: evidence from molecular biology. In Darwin Today (eds E. Geissler and W. Scheler), pp. 95-110. Akademie-Verlag, Berlin. Grantham, R., Gautier, C., Gouy, M., Jacobzone, M., and Mercier, R. (1981). Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acids Res. 9, r43-74. Grantham, R., Gautier, C. and Gouy, M., Mercier, R., and Pavd, A. (1980b). Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 8, r49-62. Grantham, R., Greenland, T., Louail, S., Mouchiroud, D., Prato, J. L., Gouy, M., and Gautier, C. (1985). Molecular evolution of viruses as seen by nucleic acid sequence study. Bull. Inst. Pasteur, 83, 95-148. Grantham, R. and Perrin, P. (1985). Tentative de modelisation des sequences de genes hautement exprimes: rapport sur l'avancement des travaux. Rapport C.N.R.S. du 1 Novembre 1985. Grantham, R. and Perrin, P. (1986). AIDS virus and HTLV-1 differ greatly in codon choices. Nature, 319, 727-8. Grosjean, H., Sankoff, D., Min Jou, W., Fiers, W., and Cedergren, R. (1978). Bacteriophage MS2 RNA: a correlation between the stability of the codon: anticodon interaction and the choice of codewords. J. Mol. Evol. 12, 113-9. Grosjean, H. and Fiers, W. (1982). Preferential codon usage in prokaryotic genes: the optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes. Gene, 18, 199-209. Hamada, H., Petrino, M. G., and Kakunaga, T. (1982). A novel repeated element with Z-DNA-forming potential is widely found in evolutionary diverse eukaryotic genomes. Proc. Nat. Acad. Sci. USA, 79, 6465-6469. Hedrick, S. M., Nielsen, E. A., Kavaler, J., Cohen, D. I., and Davis, M. M. (1984). Sequence relationships between putative T-cell receptor polypeptides and immunoglobulins. Nature, 308, 153-8. Herr, W. (1984). Nucleotide sequence of AKV murine leukaemia virus. J. Virol. 49, 471-478. Ikemura, T. (1981). Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol. 151, 389-409. Ikemura, T. (1982). Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. J. Mol. Biol. 158, 573-97. Ikemura, T. (1985). Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol. Evol. 2, 13-34. Ikemura, T. and Ozeki, H. (1983). Codon usage and transfer RNA contents: organism-specific codon choice patterns in reference to the isoacceptor contents. Cold Spring Harbor Symp. Quant. Biol. . 47, 1087-1097. Jerne, N. K. (1985). The generative grammar of the immune system. Science, 229, 1057-1059. Johnston, B. H. and Rich, A. (1985). Chemical probes of DNA conformation: detection of Z-DNA at nucleotide resolution. Cell, 42, 713-724. Kabat, E. A., Wu, T. T., Bilofsky, H., Reid-Miller, M., and Perry, H. (1983). In Sequences of Proteins of Immunological Interest. U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health. Kam, J., Brenner, S., and Barnett, L. (1983). Protein structural domains in the Caenorhabditis elegans unc-54 myosin heavy chain gene are not separated by introns. Proc. Nat. Acad. Sci. USA, 80, 4253-4257. Klass, M. R., Kinsley, S., and Lopez, L. C. (1984). Isolation and characterization of a sperm-specific gene family in the nematode Caenorhabditis elegans. Mol. Cell. Biol. 4, 529-537. Kramer, J. M., Cox, G. N., and Hirsh, D. (1982). Comparisons of the complete sequences of two collagen genes from Caenorhabditis elegans. Cell, 30, 599-606. Li, W. H., Luo, C. C., and Wu, C. I. (1985). Evolution of DNA sequences. In Molecular evolutionary genetics (ed. R. J. MacIntyre), pp. 51-65. New York: Plenum Press. Marini, M. and Mushinski, J. F. (1979). Transfer ribonucleic acids from eleven immunoglobulin secreting mouse plasmacytomas. Constant and variable chromatographic profiles compared with the myeloma protein sequences. Biochim. Biophys. Acta, 562, 252-270. Miyata, T., Hayashida, H., Yasunaga, T., and Hasegawa, M. (1979). The preferential codon usages in variable and constant region of immunoglobulin genes are quite distinct from each other. Nucleic Acids Res. 7, 2431-2437. N'Guyen, C., Sodoyer, R., Trucy, J., Strachan, T., and Jordan, B. R. (1985). The HLA-AW24 gene: sequence, surroundings and comparison with the HLA-A2 and HLA-A3 genes. Immunogenetics, 21, 479-89. Nisonoff, A., Hopper, J. E., and Spring, S. B. (1975). Human immunoglobulins. In The Antibody Molecule, pp. 86-137. Academic Press, London. Nordheim, A. and Rich, A. (1983). The sequence (dc-dA)n-(dG-dT)n forms left-handed Z-DNA in negatively supercoiled plasmids. Proc. Nat. Acad. Sci. USA, 80, 1821-1825. Patten, P., Yokota, T., Rothbard, J., Chien, Y. H., Arai, K. I., and Davis, M. M. (1984). Structure, expression and divergence of T-cell receptor P-chain variable regions. Nature, 312, 40-6. Perlmutter, R. M., Crews, S. T., Douglas, R., Sorensen, G., Johnson, N., Nivera, N., Gearhart, P 0 J., and Hood L. (1984). The generation of diversity in phosphorylcholine-binding antibodies. Adv. Immunol. 35, 1-37. Perrin, P. (1984). Coding strategy differences between constant and variable segments of immunoglobulin genes. Nucleic Acids Res. 12, 5515-37. Perrin, P. and Grantham, R. (1986). Avoidance of base runs in switch regions of immune system genes. (Submitted.) Ratner, L., Haseltine, W., Patarca, R., Livak., K. J., Starcich, B., Josephs, S. F., Doran, E. R., Rafalsky, J. A., Whitehorn, E. A., Baumeister, K., Ivanoff , L., Petterway, S. R., Jr, Pearson, M. L., Lautenberger, J. A., Papas, T. S., Ghrayeb, J., Chang, N. T., Gallo, R. C., and Wong-Staal, F. (1985).. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature, 313, 277-84. Robertson, M. (1985). The present state of recognition. Nature, 317, 768-771. Roe, B. A., Ma, D. P., Wilson, R. K., and Wong, J. F. H. (1985). The complete nucleotide sequence of the Xenopus laevis mitochondrial genome. J. Biol. Chem. . 260, 9759-74. Rogers, J. (1983). CACA sequences - the ends and the means? Nature, 305, 101-2. Sablitzky, F., Wildner, G., and Rajewsky, K. (1985). Somatic mutation and clonal expansion of B cells in an antigen-driven immune response . Embo. J. 4, 345-50. Saccone, C., Cantatore, P., Gadaleta, G., Gallerani, R., Lanave, C. , Pepe, G., and Kroon, A. M. (1981). The nucleotide sequence of the large ribosomal RNA gene and the adjacent tRNA genes from rat mitochondria. Nucleic Acids Res. 9, 4139-48. Sagata, N., Yasunaga, T., Tsuzuku-Kawamura, J., Ohishi, K., Ogawa, Y., and Ikawa, Y. (1985). Complete nucleotide sequence of the genome of bovine leukemia virus: its evolutionary relationship to other retroviruses. Proc. Nat. Acad. Sci. USA, , 82, 677-81. Saito, H., Kranz, D. M., Takagaki, Y., Hayday, A. C., Eisen, H. N., and Tonegawa, S. (1984). Complete primary structure of a heterodimeric T-cell receptor deduced from cDNA sequences. Nature, 309, 757-762. Schwartz, D. E.. Tizard, R., and Gilbert, W. (1983). Nucleotide sequence of Rous sarcoma virus. Cell, 32, 853-69. Seiki, M., Hattori, S., Hirayama, Y., and Yoshida, M. (1983). Human adult T-cell leukaemia virus: complete nucleotide sequence of the provirus genome integrated in leukaemia cell DNA. Proc. Nat. Acad. Sci. USA, 80, 3618-3622. Shepherd, J. C. W. (1982). From primeval message to present-day gene. Cold Spring Harbor Symp. Quant. Biol. 46, 1099-1108. Shepherd, J.C. W. (1984). Fossil remnants of a primeval genetic code in all forms of life? Trends Biochem. Sci. 9, 8-10. Shinnick, T. M., Lerner, R. A., and Sutcliffe, J. G. (1981). Nucleotide sequence of Moloney murine leukaemia virus. Nature, 293, 543-548. Sodoyer, R., Damotte, M., Delovitch, T. L.,. Trucy, J., Jordan, B. R., and Strachan, T. (1984). Complete nucleotide sequence of a gene encoding a functional human class I histocompatibility antigen (HLA-CW3). Embo J. 3, 879-85. Sonigo, P., Alizon, M., Staskus, K., Klatzmann, D., Cole, S., Danos, O., Retzel, E., Triollais, P., Haase, A., and Wain-Hobson, S. (1985). Nucleotide sequence of the Visna lentivirus: relationship to the AIDS virus. Cell, 43, 369-382. Spieth, J., Denison, K., Zucker, E. and Blumenthal, T. (1985). The nucleotide sequence of a nematode vitellogenin gene. Nucleic Acids Res. 13, 7129-38. Sprinzl, M., Moll, J., Meissner, F., and Hartmann, T. (1985). Compilation of tRNA sequences. Nucleic Acids Res. 13, rl-49. Steinmetz, M. (1984). Structure, function and evolution of the major histocompatibility complex of the mouse. Trends Biochem. Sci. 9, 224-6. Storb, U. and Arp, B. (1983). Methylation patterns of immunoglobulin genes in lymphoid cells: correlation of expression and differentiation with undermethylation. Proc. Nat. Acad. Sci. USA, 80, 6642-6646. Strachan, T., Sodoyer, R., Damotte, M., and Jordan, B. R. (1984). Complete nucleotide sequence of a functional class I gene, HLA-A3: implications for the evolution of HLA genes. Embo J. 3, 887-894. Temin, H. M. (1985). Reverse transcription in the eukaryotic genome: retroviruses, pararetroviruses, retrotransposons and retrotranscripts. Mol. Biol. Evol. 2, 455-68. Tonegawa, S. (1983). Somatic generation of antibody diversity. Nature, 302, 575-81. Tykocinski, M. L. and Max, E. E. (1984). CG dinucleotide clusters in MHC genes and in 5' demethylated genes. Nucleic Acids Res. 12, 4385-4396. Weiss, E., Golden, L., Zakut, R., Mellor, M., Fahmer, K., Kvist, S., and Flavell, R. A. (1983). The DNA sequence of the H-2Kb gene: evidence for gene conversion as a mechanism for the generation of polymorphism in histocompatibility antigens. Embo J. 2, 453-62. Wolf, S. F. and Migeon, B. R. (1985). Clusters of CpG dinucleotides implicated by nuclease hypersensitivity as control elements of housekeeping genes. Nature, 67, 449-53.
It would be nice to know more about Richard Grantham's life. His friend, Timothy Greenland, tells me RG died in 2009. I can find no obituary notices. The US Social Security Death Index lists a Richard L. Grantham as having been born April 9 1922 and as having died July 28 2009. This seems about right, but there are many RG's out there. If anyone has information on RG that they would be willing to share, please contact me. It would be nice to know more about the founder of Evolutionary Bioinformatics (EB). Donald Forsdyke
After some correspondence, I finally met Richard at the 2000 Ischia workshop on "Neutralism and Selectionism" (Click Here). At that time he was appeared well and we had a splendid discussion followed up by even more correspondence. For many years Richard was deeply concerned to find some way to remedy the environmental degradation of our planet and sought out Thomas Goreau with whom there was a long correspondence. He and Timothy Greenland have published their reminiscences of Richard in the forward to a book, where he is hailed as "the father of geotherapy:" Geotherapy. Innovative Methods of Soil Fertility Restoration, Carbon Sequestration, and Reversing CO2 Increase. (Goreau TJ, Larson RW, Campe J, editors) CRC Press, 2015 (Taylor & Francis Group) pp. ix-xi.
Richard Grantham: Father of Geotherapy, 1922�2009 It must have been in the late
1970s when I first met Richard Grantham. He came to give a lecture at the
research laboratory where I worked and described how he had used a
computer analysis of the genetic sequences present in a virus capable of
causing cancer to identify a gene that had its origins in the virus's
host, not in the ancestors of the virus. I was fascinated. Some years
before, I had attempted to develop computer programs for biological
problems, although I am a dreadful mathematician, and I asked him if, with
the consent of my director, I could come and collaborate with his group
one day a week. He agreed, and this arrangement lasted throughout our
working careers. His group developed many of the basic computer techniques
employed today for the accession and analysis of genetic sequences - it
was an exciting time and I learned much. A lot of the time was spent
discussing projects and future research directions together in English,
because although Richard, like me, was very fluent in French (we both had
French wives), we sometimes found it useful to revert to our native
language for the more abstruse concepts. As time passed, these discussions
took a more philosophical turn and Richard became progressively more
concerned with the degradation of the biosphere and the impact of humanity
on the natural world. The
contributions that Richard and his colleagues made to the dawning science
of the molecular biology of genetic sequences, and their relevance to
evolutionary processes, were many and varied. He is most often remembered
in the context of his genome hypothesis, which allowed that the DNA
sequence in the gene itself carried to some degree a signature
characteristic of the species (or species group) from which that gene
derived. Classically at that time, it was considered that Darwinian
selection could only operate at the level of proteins because the genes
themselves were only visible to selection pressures through their
expressed products. However, most of the amino acids that compose proteins
can be specified by more than one sequence of three nucleotides (codon) in
the genetic DNA. Richard realized that the different alternative codons
were not used randomly in the genes from different species and that there
was a pattern of use that appeared to be common to different genes from
the same species. It must be realized that, for a paper published in 1980
(1), the total number of sequences available in his pioneering database
was 160. As of April 2011, GenBank contained 135,440,924 sequences! It is
not surprising that his initial insight and analysis have now been greatly
developed by later work, but it remains amazingly relevant today. For a
deeper view of his contributions, I recommend a visit to
Grantham's
Genome Hypothesis (queensu.ca) where Donald Forsdyke
presents some of his works. Professor Forsdyke's own contributions to this
important field may be found on the same website. During our
discussions, we found that we had both read and been affected by Rachel
Carson's The Silent Spring and we quickly discovered Aldo Leopold's A Sand
County Almanac: And Sketches Here and There, then the influential books by
Van Rensselaer Potter (see References). These convinced Richard that he
should concern himself actively with the problems of human impact on the
natural world, particularly the concept to global bioethics. Incidentally,
we both deplored the hijacking of the simple term bioethics by the medical
community - despite clear priority for the term for ecological concerns.
Among other important inputs, I would mention especially John Rawls' A
Theory of Justice and Julia Annas' The Morality of Happiness, which helped
us consider the knotty problems of the impact of bioremediation and the
instauration of a sustainable lifestyle on a the real human population. Despite
the hundreds of hours of discussion and exchange with Richard, I realized
that I would never get more than a glimpse of the complex and profound
person I was privileged to encounter. We became friends, but I never knew
much of his background or personal life. He had served as a bomber pilot
during World War II in Italy and returned to Europe to study evolutionary
biology at the University of Montpelier in the 1960s. I learned little
about his youth in central California or of his family. Any diversion in
our conversations in those directions was quickly brought back to the
central theme of his projects and philosophical analyses. On occasion we
would find time to walk through the woods together - sometimes hunting
mushrooms, and always alert for whatever wildlife we should chance to
meet. His knowledge was wide and eclectic and it was a joy to accompany
him on these diversions. Richard
corresponded quite extensively with Van Rensselaer Potter on themes of
mutual interest, and I had the pleasure of participating in their
exchanges. I think that both of them appreciated finding someone of
similar stature to converse with, and Richard met Van on a rare visit to
the United States. He brought back some very helpful insights and several
pleasant reminiscences. His mind turned progressively to a very practical
problem: The damage is done - how can we mitigate its effects? To this
end, he imagined grandiose schemes like the irrigation of North African
dessert areas with desalinated water to reconstitute the forests that
existed there not so long ago (at least in geological timescales).
Geotherapy became his watchword. The earth is already very sick, and
heroic remedies are necessary to give even a chance of the survival of our
patient. As an evolutionary biologist, he was only too aware of the perils
of an overreaching species. It was clear to both of us that the carrying
capacity of the planet for Homo sapiens had been exceeded for a long
while. Richard did not despair of a downturn to more supportable human
numbers and balanced survival, given that we could buy time for the
message to be heard and understood by our species as a whole. I confess to
being less optimistic. We both gave much time and thought to possible
remedial approaches - which are, indeed, both marginally possible and
vitally urgent - but the scale of the problem and the intransigence of
human politics remained discouraging. We both feared
that the natural solution would be the elimination, probably through
disease, of much of the human population - and that our experience of
studies of such events in other species suggested that the most frequent
outcome is extinction and not simple population reduction! Humanity is
perhaps the first species to be able to see the consequences of its
proliferation, even if dimly, but will it be able to act on those
warnings? We both agonized on the unfolding of the AIDS epidemic and
contributed to studies of the virus and its consequences. We have perhaps
postponed that threat. Then came other novel pathogens, and more will
certainly come. Each may be the last. The question is simply one of
survival, not of preservation of comforts, but life or death. Will we
measure up? He and Professor Potter jointly http://www.thewip.net/contributors/2010/05/geotherapy_artist_mara_haselti.html - I am not sure what Richard's reaction would have been, but knowing his deep interest in, and appreciation for, matters artistic, I am sure he would have been intrigued! References Donald Forsdyke
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||






![Base composition [C+G)%] of human and viral genes](images/granth06.gif)