Genomic
conflict settled in favour of the species rather than of the gene at extreme GC%
values
Shang-Jung Lee, James R Mortimer and Donald R Forsdyke
Applied
Bioinformatics (2004) 3, 219-228
![]()
Species differences in GC% among prokaryotes
Genic differences in GC% within a bacterium
Switch from species to genic demands
Role of GC% in species selection (isolation)
Role of GC% in Gene Selection (Isolation)
![]()
|
Abstract
:
Wada and coworkers have shown that, whether prokaryotic or eukaryotic, each gene has a "homostabilizing propensity" to adopt a relatively uniform GC%. Accordingly, each gene can be viewed as a "microisochore" occupying a discrete GC% niche of relatively uniform base composition amongst its fellow genes. Although first, second and third codon positions usually differ in GC%, each position tends to maintain a uniform, gene-specific, GC% value. Thus, within a genome, genic GC% values can cover a wide range. This is most evident at third codon positions, which are least constrained by amino acid-encoding needs. In 1991 Wada and coworkers further noted that, within a phylogenetic group, genomic GC% values can also cover a wide range. This is again most evident at third codon positions.
Wada described the context-independence of plots of different codon position GC% values against total GC% as a "universal" characteristic. Several studies relate this to recombination. We have confirmed that third codon positions usually relate more to the genes that contain them than to the species. However, in genomes with extreme GC% values (low or high) third codon positions tend to maintain a constant GC%, thus relating more to the species than to the genes that contain them. Genes in an extreme GC% genome collectively span a less wide GC% range, and mainly rely on first and second codon positions for differentiation as "microisochores." Our results are consistent with the view that differences in GC% serve to recombinationally isolate both genome sectors (facilitating gene duplication) and genomes (facilitating genome duplication; e.g. speciation). In intermediate GC% genomes, conflict between the needs of the species and the needs of individual genes within that species is minimal. However, in extreme GC% genomes there is a conflict, which is settled in favour of the species (i.e. group selection), rather than of the gene (genic selection). Keywords: base composition - codon position - gene duplication - speciation |
|
In
most definitions of "gene" there is a loose or explicit reference to function. However,
Williams defined a gene as any DNA segment that can persist for enough
generations to serve as a unit of natural selection. This required that it not
be disruptable by recombination (Williams 1966).
For Williams, preservation against disruption was achieved by one agency,
natural selection. Shortly thereafter, evidence began to appear that another
agency, base composition, might be involved in the preservation of gene-sized
DNA segments. Prior to modern sequencing technologies, small segments of relatively uniform GC% were demonstrated in the genomes of prokaryotes and their viruses. Following extensive hydrodynamic sheering to produce subgenome-sized duplex fragments, the 48 kb duplex genome of phage lambda was resolved into six segments of differing densities on salt gradients. The segments were "homogeneous internally and the boundaries between segments rather sharp." The densities were related to the average GC% values of the segments, GC-rich DNA being denser than AT-rich DNA. It was noted that, if "average nucleotide composition can itself influence DNA function -- direct selection for the given composition is possible," so that demarcation of segments of relative GC% uniformity might have occurred in order "not to encourage recombination within functional units" (Skalka et al 1968). The phage lambda genome was further resolved into thirty four "gene sized" segments by thermal denaturation spectrophotometry, which is very sensitive to GC% differences (Vizard and Ansevin 1976). Similar thermal denaturation studies led to the observation by Wada et al. (1976) that each gene within a prokaryotic genome has a "homostabilizing propensity," so that "every base in a codon seems to work cooperatively towards realizing the gene's characteristic value of (G + C) content." This was later buttressed by direct sequence analyses (Bibb et al 1984; Wada and Suyama 1985). It was found "hard, if not impossible, to believe" that the homostabilizing regions reflect a fundamental characteristic of the genetic code itself. Rather the regions must play "an important part somewhere in the biological process within which the DNA is closely related -- . From the size of the homostability region, recombination might be one possible process --" (Wada et al 1976). Further thermal denaturation studies showed that the "gene homostabilizing propensity" also extended to eukaryotes and their viruses (Suyama and Wada 1983; Wada and Suyama 1986). Prior to modern sequencing technologies, segments of relatively uniform GC% were also identified in many eukaryotic genomes, and named "isochores" (Clay et al 2003). This entailed the sheering of duplex DNA into fragments around 300 kb, which were again separated on salt gradients on the basis of density differences. This methodology made it inevitable that isochores (Greek: iso = equal; choros = group) would initially be defined in terms of size, as well as of their distinctive base compositions, and would be perceived as being eukaryote-specific. Being large, they often contained many genes of which bases at third codon positions and sometimes the encoded amino acids (mainly determined by bases at first and second codon positions) were reflective of isochore GC%. It is now evident that eukaryotic genes, like prokaryotic genes, pass from generation to generation in a "coat" of a particular GC% "colour," depending on their isochore. Protein-encoding characteristics (largely determined by first and second codon positions), and the GC% "colour" (largely determined by third codon positions), are coinherited. For example, the human α-globin gene resides in a high GC% isochore and has a GC-rich "coat." The human β-globin gene, encoding a similar amino acid sequence, resides in a low GC% isochore and has an AT-rich "coat" (Efstratiadis et al 1980). Again, the possibility of a function related to recombination is entertained. It is proposed that location in an isochore of distinct GC% acts as "a constraint to avoid recombination with related sequences elsewhere in the genome" (Matsuo et al 1994). Thus, "recombination explains isochores" (Montoya-Burgos et al 2003; Iwase et al 2003). In this light, every gene, be it prokaryotic or eukaryotic, can now be viewed as a "microisochore." Classical isochores can be viewed as collections of "microisochores" within a region of relatively uniform GC%. Classical isochores are not detected in prokaryotic genomes because the microisochores are not generally collected into large regions of relatively uniform GC% (Nomura et al 1987; Sueoka 1992; Li 2001). Just as individual isochores differ from other isochores within a genome with respect to GC%, so individual genomes differ from other genomes within a phylogenetic group with respect to GC%. If GC% differences between isochores have the potential to confer protection against inadvertent recombination on functional intragenomic units (e.g. genes), so GC% differences between species should have the potential to confer protection against inadvertent recombination on functional genomic units (e.g. species; Forsdyke 1994; 1995; Forsdyke and Mortimer 2000). This raises the possibility of conflict between genic GC% and genomic GC%, a conflict that might be particularly apparent in genomes of extreme GC%. Genome sequences now being available from many species, we here present a study of genic codon position GC% values in extreme GC% genomes. We show that, whereas GC% can often serve both genic and genomic (e.g. species) masters, in extreme GC% genomes there is indeed a conflict and this is resolved in favour of the species. |
|
Base
compositions at different codon positions were calculated from codon usage
tables using programs written in Perl and programs written for Microsoft Excel
in Microsoft Visual Basic for Applications (Mortimer and Forsdyke 2003). CUTG ("Codon Usage Tables from GenBank") are derived from
the available protein-encoding sequences of a species and are automatically
updated with each new GenBank release (Nakamura
et al 2000). However, there is no automatic screening for redundancies, so this
can create problems with some applications. For example, different isolates of
the same gene may be deposited, but scored as independent genes. Furthermore,
there is bias because certain genes are chosen for sequencing before others.
Nevertheless, exclusive reliance on fully sequenced genomes disregards the great
range and quantity of information available from incompletely sequenced genomes.
Happily, some of our previous comparisons of the different codon positions of
incompletely sequenced genomes (Lambros et al 2003) have been confirmed by
recent independent studies with much fewer, but completely sequenced, genomes
(Paz et al 2004). In the present work we excluded species with less than twenty individual protein-encoding sequences when calculating the slopes of codon position plots (Figs. 2-5), but found further curation was unnecessary. Thus, for the bacterium Candidatus Carsonella ruddii (Fig. 2a) we obtained the base compositions for different codon positions from each of 87 open reading frames (ORFs) corresponding to a diversity of protein-encoding genes (e.g. alkyl hydroperoxide reductase, ATP synthase subunits, gidA cell division protein, ornithine transcarbamoylase, various ribosomal proteins, various RNA polymerase subunits, transaldolase, tufA and other elongation factors, various tRNA synthetases). ORFs differing by two or less bases from the first ORF of a particular size class (sometimes different isolates of the same gene), were eliminated to produce 48 ORFs. The slope parameters were virtually the same as those shown in Fig. 2a. Small errors of this nature should have been adequately compensated for statistically by the large quantity of sequences that this approach made available (summarized in Fig. 5). |
|
Species differences in GC%
among prokaryotes |
|
|
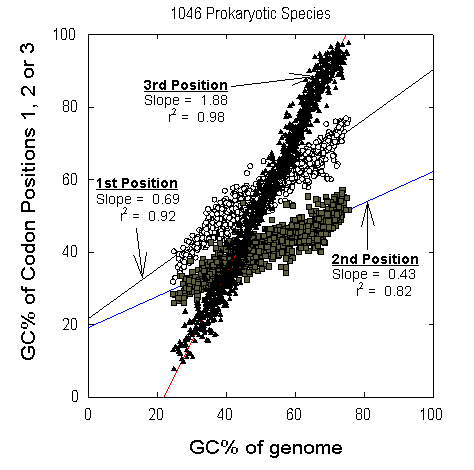
Figure 1 Relative contributions of different codon positions to species GC% in each of the 1046 prokaryotic species (eubacteria, archaebacteria) represented in GenBank in September 2000 by four or more genes. For each species there are 3 values corresponding to the average GC% of first codon positions (open circles), the average GC% of second codon positions (grey squares), and the average GC% of third codon positions (black triangles), for the set of all sequenced genes of the species. These three values are plotted against an estimate of the GC% for the entire genome of the species, derived from the sum of the base compositions of the coding regions of all sequenced genes. Slope and adjusted r2 values of the first order linear regression lines are shown for each position. This figure is reproduced from Mortimer and Forsdyke (2003). |
|
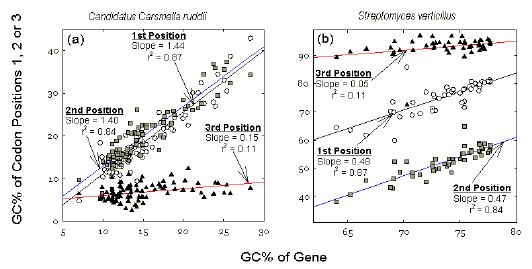
Genic differences in GC% within a bacterium Figure 2 shows plots for genes within two genomes of extreme average GC% values (low and high). Average GC% values of different codon positions in each sequenced gene are plotted against the average GC% value of that gene. These average genic GC% values are distributed over the 5-30% range in the low GC% genome (see abscissa of Fig. 2a), and over the 60-80% range in the high GC% genome (see abscissa of Fig. 2b). Thus, within a genome individual genes are differentiated from each other with respect to their average GC% values. A major role of third codon positions in reflecting the extreme GC% values of the genomes is evident. Figure 2a shows that, in a low GC% genome, third codon positions are almost independent of the average GC% of the genes that contain them. Thus, the slope of the plot for third codon positions is close to zero. Whatever the gene, its third codon position GC% value is about 7%. Similarly, in a high GC% genome, whatever the gene, its third codon position GC% value is around 94% (Fig. 2b). Thus, the average GC% of a particular gene (i.e. its differentiation from other genes) is largely determined by the GC% of first and second codon positions. |
|
|
|
Figure
2 Within-species variation in contributions to the GC% of
individual bacterial genes by different codon positions for (a) a low genomic
GC% species (Candidatus Carsonella ruddii; 15.6% GC; 87 sequenced genes),
and (b) a high genomic GC% species (Streptomyces verticillus; 75.0% GC;
48 sequenced genes).
Each point represents the average GC% value for a
particular codon position in all the codons of a particular gene. These three
values are plotted against an estimate of the GC% for the gene, derived from the
base composition of the protein-encoding region (open reading frame). Data are
from the December 2001 release of GenBank. Symbols for codon positions are as in
Figure 1. |
|
On this basis, one might predict that, in the case of a bacterial species of intermediate genomic GC%, all genes within the genome would again have relatively constant third codon position GC% values, so that the slope of the third codon position plot would again be low. In fact, as noted by Wada, Suyama and Hanai (1991), within-species genic plots (e.g. Fig. 3) are very similar to between-species genomic plots (Fig. 1). The plots are "universal," being independent of context (genes or species). In contrast to bacteria with extreme genomic GC% values (Fig. 2), in a bacterium with intermediate genomic GC% values the third codon position shows the greatest change in GC% (i.e. the slope of the plot is greater than the slopes of the plots for the first and second codon positions). |
|
|
| Figure 3 Within-species variation in contributions by different codon positions to the GC% of 4290 genes of an intermediate genomic GC% species, (E. coli K12; 51.8% GC). Each point represents the average GC% value for a particular codon position in all the codons of a particular gene. Data are from the December 2001 release of GenBank. |
|
Thus, in intermediate GC% genomes the third codon position appears to amplify within a gene either a downward pressure towards low GC% by exceeding the first and second codon positions in its AT-richness, or an upward pressure towards high GC% by exceeding the first and second codon positions in its GC-richness. Accordingly, genes are highly differentiated, with average GC% values covering a range from about 25% to 65% (see abscissa of Fig. 3). All codons within a particular gene tend towards a distinct GC% ("gene homostabilizing propensity;" Wada and Suyama 1985). Genic differences in GC% within a eukaryote Similar results are found in eukaryotes (e.g. D'Onofrio and Bernardi 1992). We examined the GenBank gene collections for invertebrates and plants, which include a wide range of organisms differing in their average genomic GC%. Figures similar to Figure 1 were obtained when genomes within these collections were compared (data not shown). As an example of a eukaryotic genome of extreme GC% (low), Figure 4a shows plots for genes on the second chromosome of the malarial parasite, Plasmodium falciparum (Gardner et al 1998). The plots for this invertebrate resemble those obtained with bacterial species of low genomic GC% (e.g. Fig. 2a). As an example of a eukaryotic species of intermediate genomic GC%, Figure 4b shows plots for rice genes (Oryza sativa; Yu et al 2002). The plots resemble those obtained with bacterial species of intermediate genomic GC% (e.g. Fig. 3). |
|
|
| Figure 4 Codon position plots of genes in eukaryotes resemble those of genes in prokaryotes of the same genome GC%, as exemplified by (a) 205 genes from the second chromosome of the malaria parasite, P. falciparum (GC% = 19.7) and (b) 3111 genes of the rice species, Oryza sativa (GC% = 43.3). Data are from the December 2001 release of GenBank. |
|
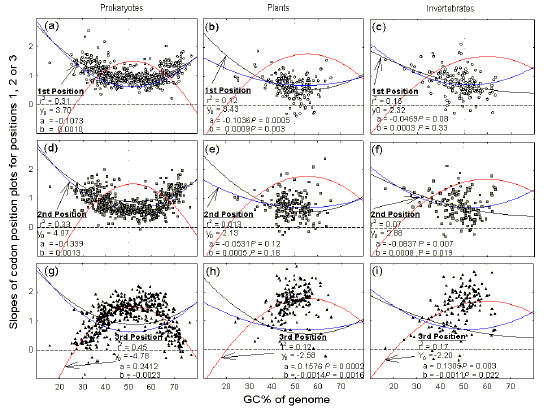
In species of extreme genomic GC%, slope values for genic first and second codon positions exceed those for genic third positions, whereas the opposite hold for species of intermediate genomic GC% (Figs. 2-4). To determine at what GC% the patterns switch, and to demonstrate the generality of the results, the three slope values from plots for each of many individual species (e.g. slope values from Figs. 2-4), were plotted against the average genomic GC% values for those species. Data for 546 prokaryotic species, 195 plant species, and 153 invertebrate species, were fitted to second order linear regressions (Fig. 5). |
|
|
|
Figure 5 Slopes of plots for
genic third codon positions are high in species of intermediate genomic GC% and
low in species of extreme genomic GC%, whereas slopes for genic first and second
codon positions show the opposite trend.
This applies to prokaryotes (a,d,g; 546 species, each with 20 or more sequenced genes), plants (b,e,h; 195 species, excluding organelles, each with 20 or more sequenced genes), and invertebrates (c,f,i; 153 species, excluding organelles, each with 20 or more sequenced genes). Slope values were fitted to second order regression curves (Y = Y0 + aX + bX2) for which there are two parameters (a and b); associated P values are shown when they are >0.0001. Symbols as in previous figures. Data from the August 2002 GenBank release. |
|
Prokaryotic species cover the widest range of average genomic GC% values, with genic third codon position slope values peaking at about 1.5 in species of intermediate genomic GC%, and declining to about zero in species of extreme genomic GC% (Fig. 5g). In contrast, genic first and second position slope values are about 0.9 in species with intermediate genomic GC%, and increase to about 1.5 in species with extreme genomic GC% (Fig. 5a, d). The curves cross at about 38% and 68% GC. Species of high genomic GC% are rare in the plant and invertebrate collections, but the same general patterns are observed. For plants, the crossing points from low genomic GC% species to intermediate genomic GC% species occur at about 32% GC (Figs. 5b,e,h). For invertebrates, the corresponding crossing points occur at about 34% (Figs. 5c,f,i). |
|
Switch from species to genic
demands We observe that species with extreme genomic GC% values (low or high) sustain these values largely by maintaining constant, gene-independent, extreme third codon position genic GC% values. However, values for first and second codon positions, that specify amino acids, are partly gene-dependent. Thus, the latter positions appear to serve both general demands of the genome (species GC%) and local demands of protein-encoding (Figs. 2, 4a). The situation changes dramatically in species with intermediate genomic GC% values. All three codon positions assume gene-dependent GC% values, and this is most evident in the case of third codon positions (Figs. 3, 4b). Accordingly, a low GC% gene has many AT-rich codons such as TTY (generic codon for phenylalanine, with Y indicating either T or C). Here TTT is preferred over TTC. On the other hand, a high GC% gene has many GC-rich codons such as GGN (generic codon for glycine, with N indicating any base). Here GGC and GGG are preferred over GGT and GGA. These codons are usually distributed relatively uniformly along the length of the gene. Thus, third positions support and extend the GC% values of first and second positions. In this way each gene in a genome of intermediate GC% has a "homostabilizing propensity" and has come to occupy a discrete GC% niche, or "microisochore," amongst its fellow genes, which collectively span a wide GC% range. So why are species with extreme genomic GC% values different? It is intuitively apparent that a sequence with only one base would have less ability to transmit information than a sequence with four bases. In fact, elementary information theory shows that information transmission potential is maximized when the four bases are present in equal proportions (Khinchin 1957; Wan and Wootton 2000). Thus, departures from 50% GC (downwards or upwards) are likely to progressively compromise the ability of genomes to transmit further information. In other words, when you are close to a wall (0% or 100% GC) there is less room to manoeuvre, so there may have to be trade-offs. The trade-off here seems to be that, at extreme genomic GC% values, third codon positions serve the information demands of a species, rather than of individual genes within a species. Note that we refer here to a genome transmitting further information. It has been argued that an extreme genomic GC% value is the result of a species-specific mutational bias of no adaptive significance (i.e. no information of importance to a gene, individual or species is conveyed). Alternatively, it has been argued that the extreme GC% value is itself information of adaptive significance. These arguments will be considered later. In species with intermediate genomic GC% values, the contribution to genic demands is shared by all codon positions, with third positions making the greatest contributions. In species with extreme genomic GC% values, third positions contribute little to genic demands. Seeming to compensate for this, first and second codon positions increase their contributions. Accordingly, the slope values for genic first and second positions in species with extreme genomic GC% values is greater than slope values for genic first and second positions in species with intermediate genomic GC% values (Fig. 5). Thus, even in species with extreme genomic GC% values, there appears to be some genic differentiation into microisochores. However, the range of this differentiation is much less than in species with intermediate genomic GC% values. It follows that the nature of the amino acids encoded by genes in species with extreme genomic GC% values is strongly influenced by GC%. In a genome such as that of P. falciparum, this constraint appears to have increased the lengths of proteins due to their incorporation of low complexity segments that serve the needs of the encoding nucleic acid, not of the encoded protein (Forsdyke 2002a, b; Xue and Forsdyke 2003). A gene differing dramatically in GC% from the average GC% of the genes in a genome might be considered to be the result of a "horizontal" gene transfer from the genome of another species of different average GC%. While this might sometimes be the case, we have shown here that such apparent "maverick" genes may actually be part of the normal distribution of genes in a genome (Figs. 2-4). Sueoka (1992) proposed that regions of relative GC% uniformity in prokaryotes could "be regarded as a simple form of isochores," Since isochores, as we here define them, are present both in prokaryotes and eukaryotes, this argues for the fundamental nature of isochores, and implies that they could have been a feature of a common ancestor of modern prokaryotic and eukaryotic lineages. Along similar lines, Sueoka (1992) considered isochores "a ubiquitous phenomenon from bacteria to mammals and that the only difference is the extent of heterogeneity between unicellular and multicellular organisms." Supporting earlier work (Nomura et al. 1987), recent studies using a recursive segmentation approach confirm that many prokaryotic genomes contain a mosaic of short polygenic segments of distinct GC% (i.e. there is within-genome differentiation of GC%; Li 2001; Bernaola-Galvan et al. 2004). However, the distributions of such polygenic segments, and of individual genes of distinct GC%, are such as to avoid the generation of classical isochores within prokaryotic genomes. Thus, when viewed on a large scale, each prokaryotic genome has a distinctive, but relatively uniform, GC%. Having earlier suggested a role in recombination (Wada et al 1976), in 1991 Wada, Suyama and Hanai drew explicit attention to the similarities between codon position plots for species within a phylogenetic group (Fig. 1) and for genes within a species (Figs. 3, 4b). They suggested that the plots "might be universal ones and the constraint parameters might have general biological meanings in relation to the DNA/RNA and protein functions". Emerging evidence suggests that the "general biological meanings" of isochores might relate to their role in a fundamental process - recombination within a genome (Montoya-Burgos et al 2003; Iwase et al 2003). To understand this, we must first consider the evidence for a role of GC% differences in another fundamental process, recombination between genomes.
Chargaff identified GC% as a species-specific base compositional parameter by direct chemical analysis of duplex DNA samples (Forsdyke and Mortimer 2000). This was later explained as due either to species-specific mutational biases (including biased gene conversion), or to classical Darwinian natural selection, or to species selection (recombinational isolation). Advocates of the mutational bias hypothesis (i.e. distinctive intrinsic directional mutational pressures from AT towards GC, or the reverse; Filipski 1990; Sueoka 1992; Sharp et al 1993; Eyre-Walker and Hurst 2001; Galtier 2003), and advocates of the natural selection hypothesis (Bernardi 2000, 2001), have argued the relative merits of their hypotheses. However, neither has addressed the species selection hypothesis (Forsdyke 1994, 1995, 1996, 1998, 1999, 2000, 2001, 2003, 2004a). Early between-genome divergence of GC% can accompany speciation (Sueoka 1961; Bellgard et al. 2001). This divergence in base composition may provide not only an early index of divergent evolution, but also a basis for the initiation of that divergence. We view GC% uniformity as a species-selective factor (i.e. species isolating factor) that, accordingly, must have arisen early in evolution. To initiate sympatric branching evolution, diverging members of a species must acquire reproductive isolation (i.e. recombinational isolation) from other members of their species (Forsdyke 2001). This they can achieve by differing in GC% (see below). If this condition does not hold, then intergenomic recombination ("blending") can occur and the identity of incipient species can be lost. By the same token, members of its allied species (the result of past successful speciation events) would exert a selection pressure on members of a species such that those that deviated in GC% values back towards those of the allied species (with which they might then be able to recombine again) are selected against (i.e. their species integrity is lost when they recombine). The best examples of this are provided by the genomes of virus species that are capable of occupying a common cytosol (Forsdyke 1996, 2001), and by studies of polyploid genomes that date back to Goldschmidt (Forsdyke 2003, 2004a).
Close matching of GC% values is likely to be necessary for a successful
homology search leading to recombination. In other words, differences in GC% are
likely to inhibit recombination. It has indeed been found that extremely small
fluctuations in GC% would suffice to change the pattern of stem-loops that
duplex DNA molecules can extrude (Forsdyke 1998). A disturbance in this pattern
would impair the loop-loop "kissing" interactions that can initiate a
paranemic homology search. Thus, it is proposed that individual species acquire
distinctive GC% values in the process of initiating recombinational isolation. Once a speciation process has begun (i.e. species have differentiated genotypically so that intergenomic recombination has been prevented), it is proposed that species differentiate phenotypically under natural selection. Then factors other than GC% can come to replace the original difference in GC% as a barrier to reproduction (i.e. a barrier to recombination). In this circumstance, third codon position GC% becomes free to adopt other roles, such as the prevention of intragenomic (e.g. intergenic) recombination (Zhang and Kishino 2004). This could involve the differentiation of genomic regions in their GC%, so generating a mosaic of microisochores and classical isochores. These regions have the potential to recombinationally isolate different parts of the genome. For example, the attempted duplication of a prototypic globin gene into a-globin and b-globin genes might have failed since sequence similarity would favour recombination between the two genes and incipient differences might then have been eliminated (by gene conversion). However, the duplication appears to have involved relocation to a different isochore with corresponding changes in GC%, so that the two genes became recombinationally isolated (Efstratiadis et al 1980). Conversely, as recently shown, a recombinationally isolated gene on being translocated to an isochore where recombination with its allele is advantageous, acquires the GC% of the host isochore. Indeed, it is proposed that "recombination explains isochores" (Montoya-Burgos et al 2003), and that "recombination suppression is somehow related to long-range mosaic structures of the genome in terms of the GC content" (Iwase et al 2003). Isochores would have arisen as a random fluctuation in base composition in a genomic region such that one product of a gene duplication was able to survive for a sufficient number of generations to allow functional differentiation of the duplicates, which might then be favoured by natural selection (Moore and Purugganan 2003). The regional base compositional fluctuation would then have "hitch-hiked" through the generations on the successful duplicate (Forsdyke and Mortimer 2000). The mutational bias hypothesis for GC% differentiation does not easily explain why individual genes are differentiated with respect to GC%. Skalka, Burgi and Hershey (1968) suggested that each local region of GC% differentiation might have "its own set of critical nucleotide sequences, each set adapted to a different mutational habit." No evidence for this highly localized "mutational habit" has since emerged. The uniformity of third codon position GC% values at extreme genomic GC percentages (high and low), is manifest as slope values close to zero (Figs. 2, 4a). Under the hypothesis of a uniform genome-wide mutational (or repair) bias, this would indicate an absence of gene-level selection that might have restrained the hypothetical genome-wide bias from affecting third codon positions. The position would, without restraint, have served this hypothetical genome-wide, species-level, bias. In this circumstance, satisfaction of protein-encoding demands (affected by local gene-level selection) would have required a flexibility at first and second codon positions that is reflected in slope values of around 1.5 (Figs. 5a, d). Yet at intermediate genomic GC percentages satisfaction of local protein-encoding demands requires a flexibility at first and second codon positions reflected in slope values of only 0.9 (Figs. 5a, d). It seems unlikely that protein-encoding demands would require less flexibility in intermediate GC% species than in extreme GC% species. The discrepancy is not easily explained in terms of a uniform genome-wide mutational bias. However, under the recombinational isolation hypothesis, at extreme genomic GC percentages local protein-encoding demands on first and second codon positions would be reflected by 0.9 slope units, as at intermediate genomic GC percentages. The local demands of genic GC-pressure (upwards and downwards) on first and second codon positions (to further increase genic microisochore differentiation) would be reflected by an extra 0.6 slope units, for a total of 1.5 slope units.
Species driven to the extremes of average GC% by pressures from allied species to recombine, cannot afford this luxury, and rely mainly on first and second codon positions to achieve genic GC% differentiation. In this case, third codon position GC% serves the demands of the species (i.e. group level selection), rather than of the genes within that species (i.e. genic level selection). This, in itself, might be of some adaptive value in the case of a parasite such as P. falciparum, since fewer barriers to recombination should facilitate antigenic variation, thus defending against host immune attack (Forsdyke 2002a, b). We propose that, in future, the term "isochore" be used to describe a general grouping consisting of "macroisochores" (i.e. the classical isochores), and "microisochores" (as described here), both being defined in terms of the uniformity of their base compositions (GC%). A fuller discussion of theoretical implications is presented elsewhere (Forsdyke 2004b).
Acknowledgements Queen's University hosts the
web-pages of DRF where full text versions of some of the cited references may be
found. |
||
|
References Bellgard M, Schibeci D, Trifonov E, Gojobori T. 2001. Early detection of G + C differences in bacterial species inferred from the comparative analysis of the two completely sequenced Helicobacter pylori strains. J Mol Evol, 53:465-468. Bernaola-Galvan P, Oliver JL, Carpena P, Clay O, Bernardi G. 2004. Quantifying intrachromosomal GC heterogeneity in prokaryotic genomes. Gene, 333: 121-133. Bernardi G. 2000. Isochores and the evolutionary genomics of vertebrates. Gene, 241:3-17. Bernardi G. 2001. Misunderstandings about isochores. Part 1. Gene, 276:3-13. Bibb MJ, Findlay PR, Johnson MW. 1984. The relationship between base composition and codon usage in bacterial genes and its use for the simple and reliable identification of protein-coding sequences. Gene, 30:157-166. Clay O, Douady CJ, Carels N, Hughes S, Bucciarelli G, Bernardi G. 2003. Using analytical ultracentrifugation to study compositional variation in vertebrate genomes. Eur Biophys J, 32:418-426. D'Onofrio G, Bernardi G. 1992. A universal compositional correlation among codon positions. Gene, 110:81-88. Efstratiadis A, Posakony JW, Maniatis T, Lawn RM, O'Connell C, Spritz RA, DeRiel JK, Forget BG, Weissman SM, Slightom JL, et al. 1980. The structure and evolution of the human β-globin gene family. Cell, 21:653-668. Eyre-Walker A, Hurst L 2001. The evolution of isochores. Nat Rev Genet, 2:549-555. Filipski J. 1990. Evolution of DNA sequence. Contributions of mutational bias and selection to the origin of chromosomal compartments. Adv Mutagenesis Res, 2:1-54. Forsdyke DR. 1994. Percentage G + C determines frequencies of
complementary trinucleotide pairs: implications for speciation. Proc Can Fed
Biol Socs, 37:152. Forsdyke DR. 1995. Relative roles of primary sequence and (G + C)% in
determining the hierarchy of frequencies of complementary trinucleotide pairs in
DNAs of different species. J Mol Evol,
41:573-581. Forsdyke DR. 1998. An alternative way of thinking about stem-loops in DNA. A case study of the G0S2 gene. J Theor Biol, 192:489-504. Forsdyke DR. 1999. Two levels of information in DNA: Relationship of Romanes' "intrinsic" variability of the reproductive system, and Bateson's "residue" to the species-dependent component of the base composition, (C+G)%. J Theor Biol, 201:47-61. Forsdyke DR. 2000. Haldane's rule: hybrid sterility affects the heterogametic sex first because sexual differentiation is on the path to species differentiation. J Theor Biol, 204:443-452. Forsdyke DR. 2001. The Origin of Species, Revisited. Montreal: McGill-Queen's University Pr. Forsdyke DR. 2002a. Selective pressures that decrease synonymous mutations in Plasmodium falciparum. Trends Parasitol, 18:411-418. Forsdyke DR, Madill, CA, Smith SD, 2002b. Immunity as a function of the unicellular state: implications of emerging genomic data. Trends Immunol, 23:575-579. Forsdyke DR. 2003. William Bateson, Richard Goldschmidt, and non-genic modes of speciation. J Biol Sys, 11:341-350. Forsdyke DR. 2004a. Chromosomal speciation: a reply. J. Theor. Biol. 230: 189-196. Forsdyke DR. 2004b. Regions of relative GC% uniformity are recombinational isolators. J. Biol. Sys.12: 261-271. Forsdyke DR, Mortimer JR. 2000. Chargaff's legacy. Gene, 261:127-137. Galtier N. 2003. Gene conversion drives GC content evolution in mammalian histones. Trends Genet, 19:65-68. Gardner MJ, Tettelin H, Carucci DJ, Cummings LM, Aravind L, Koonin EV, Shallom S, Mason T, Yu K, Fujii C, et al. 1998. Chromosome 2 sequence of the human malaria parasite, Plasmodium falciparum. Science, 282:1126-1132. Iwase M, Satta Y, Hirai Y, Hirai H, Imai H, Takahata N. 2003. The amelogenin loci span an ancient pseudoautosomal boundary in diverse mammalian species. Proc Natl Acad Sci USA, 100:5258-5263. Khinchin AI. 1957. Mathematical Foundations of Information Theory. New York: Dover Publications. Lambros, RJ, Mortimer JR, Forsdyke, DR. 2003. Optimum growth temperature and the base composition of open reading frames in prokaryotes. Extremophiles, 7:443-450. Li W. 2001. Delineating relative homogenous G + C domains in DNA sequences. Gene, 276:57-72. Matsuo K, Clay O, Kunzler P, Georgiev O, Urbanek P, Schaffner W. 1994. Short introns interrupting the Oct-2 POU domain may prevent recombination between the POU family genes without interfering with potential POU domain �shuffling' in evolution. Biol Chem Hoppe-Seyler, 375:675-683. Montoya-Burgos JI, Boursot P, Galtier N. 2003. Recombination explains isochores in mammalian genomes. Trends Genet, 19:128-130. Moore RC, Purugganan MD. 2003. The early stages of duplicate geneevolution. Proc Natl Acad Sci USA, 100:15682-15687. Mortimer JR, Forsdyke DR. 2003. Comparison of responses by bacteriophage and bacteria to pressures on the base composition of open reading frames. Applied Bioinformatics, 2:47-62. Muto A, Osawa S. 1987. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci, USA 84:166-169. Nakamura Y, Gojobori T, Ikemura T. 2000. Codon usage tabulated from the international DNA sequence databases: status for the year 2000. Nucleic Acids Res, 28:292. Nomura M, Sor F, Yamagishi M, Lawson M. 1987. Heterogeneity of GC content within a single bacterial genome and its implications for evolution. Cold Spring Harb Symp Quant Biol, 52:658-663. Paz A, Mester D, Baca I, Nevo E, Korol A. 2004. Adaptive role of increased frequency of polypurine tracts in mRNA sequences of thermophilic prokaryotes. Proc Natl Acad Sci USA, 101:2951-2956. Sharp PM, Stenico M, Peden JF, Lloyd AT. 1993. Codon usage: mutational bias, translation selection, or both? Biochem Soc Trans, 21:835-841. Skalka A, Burgi E, Hershey AD. 1968. Segmental distribution of nucleotides in the DNA of bacteriophage lambda. J Mol Biol, 34:1-16. Sueoka N. 1961. Compositional correlation between deoxyribonucleic acid and protein. Cold Spring Harb Symp Quant Biol, 26:35-43. Sueoka N. 1992. Directional mutation pressure, selective constraints, and genetic equilibria. J. Mol. Evol. 34:95-114. Suyama A, Wada A. 1983. Correlation between thermal stability maps and genetic maps of double-stranded DNAs. J Theor Biol, 105:133-145. Vizard DL, Ansevin AT. 1976. High resolution thermal denaturation of DNA: thermalites of bacteriophage DNA. Biochemistry, 15:741-750. Wada A, Suyama A. 1985. Third letters in codons counterbalance the (G + C) content of their first and second letters. FEBS Lett, 188:291-294. Wada A, Suyama A. 1986. Local stability of DNA and RNA secondary structure and its relation to biological functions. Prog Biophys Molec Biol, 47:113-157. Wada A, Suyama A, Hanai R. 1991. Phenomenological theory of GC/AT pressure on DNA base composition. J Mol Evol, 32:374-378. Wada A, Tachibana H, Gotoh O, Takanami M. 1976. Long range homogeneity of physical stability in double-stranded DNA. Nature, 263:439-440. Wan H, Wootton JC. 2000. A global compositional complexity measure for biological sequences: AT-rich and GC-rich genomes encode less complex proteins. Comput Chem, 24:71-94. Williams GC. 1966.
Adaptation and Natural Selection. Princeton: Princeton University Pr. Yu J, Hu S, Wang J, Wong GK-S, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science, 296:79-92. Zhang Z, Kishino H. 2004. Genomic background drives the divergence of duplicated Amylase genes at synonymous sites in Drosophila. Mol Biol Evol, 21:222-227.
End Note on Akiyoshi Wada (October 2014)
End Note on Maladaptation (April 2019)
|
![]()
Go to Bioinformatics Index Click Here
Go to Home Page Click Here
Abstracts: Toronto 2003, Boston 2004 Click Here
![]()
This page was placed on the Internet in December 2004 and was last edited 08 Nov 2020 by Donald Forsdyke